Ribose 5-phosphate
Ribose 5-phosphate (R5P) is both a product and an intermediate of the pentose phosphate pathway. The last step of the oxidative reactions in the pentose phosphate pathway is the production of ribulose 5-phosphate. Depending on the body's state, ribulose 5-phosphate can reversibly isomerize to ribose 5-phosphate. Ribulose 5-phosphate can alternatively undergo a series of isomerizations as well as transaldolations and transketolations that result in the production of other pentose phosphates as well as fructose 6-phosphate and glyceraldehyde 3-phosphate (both intermediates in glycolysis).
 | |
| Names | |
|---|---|
| IUPAC name
(2,3,4-Trihydroxy-5-oxo-pentoxy)phosphonic acid | |
| Identifiers | |
3D model (JSmol) |
|
| ChEBI | |
| ChemSpider | |
| ECHA InfoCard | 100.022.101 |
| MeSH | ribose-5-phosphate |
PubChem CID |
|
| UNII | |
CompTox Dashboard (EPA) |
|
| |
| |
| Properties | |
| C5H11O8P | |
| Molar mass | 230.110 |
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
| Infobox references | |
The enzyme ribose-phosphate diphosphokinase converts ribose-5-phosphate into phosphoribosyl pyrophosphate.
Structure
R5P consists of a five-carbon sugar, ribose, and a phosphate group at the five-position carbon. It can exist in open chain form or in furanose form. The furanose form is most commonly referred to as ribose 5-phosphoric acid.[1]
Biosynthesis
The formation of R5P is highly dependent on the cell growth and the need for NADPH (Nicotinamide adenine dinucleotide phosphate), R5P, and ATP (Adenosine triphosphate). Formation of each molecule is controlled by the flow of glucose 6-phosphate (G6P) in two different metabolic pathways: the pentose phosphate pathway and glycolysis. The relationship between the two pathways can be examined through different metabolic situations.[2]
Pentose Phosphate Pathway

R5P is produced in the pentose phosphate pathway in all organisms.[2] The pentose phosphate pathway (PPP) is a metabolic pathway that runs parallel to glycolysis. It is a crucial source for NADPH generation for reductive biosynthesis[3] (e.g. fatty acid synthesis) and pentose sugars. The pathway consists of two phases: an oxidative phase that generates NADPH and a non-oxidative phase that involves the interconversion of sugars. In the oxidative phase of PPP, two molecules of NADP+ are reduced to NADPH through the conversion of G6P to ribulose 5-phosphate (Ru5P). In the non-oxidative of PPP, Ru5P can be converted to R5P through ribose-5-phosphate isomerase enzyme catalysis[4].
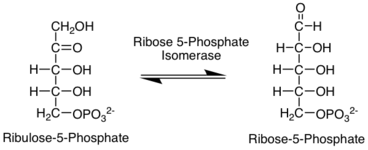
When demand for NADPH and R5P is balanced, G6P forms one Ru5P molecule through the PPP, generating two NADPH molecules and one R5P molecule.[2]
Glycolysis
When more R5P is needed than NADPH, R5P can be formed through glycolytic intermediates. Glucose 6-phosphate is converted to fructose 6-phosphate (F6P) and glyceraldehyde 3-phosphate (G3P) during glycolysis. Transketolase and transaldolase convert two molecules of F6P and one molecule of G3P to three molecules of R5P.[2] During rapid cell growth, higher quantities of R5P and NADPH are needed for nucleotide and fatty acid synthesis, respectively. Glycolytic intermediates can be diverted toward the non-oxidative phase of PPP by the expression of the gene for pyruvate kinase isozyme, PKM. PKM creates a bottleneck in the glycolytic pathway, allowing intermediates to be utilized by the PPP to synthesize NADPH and R5P. This process is further enabled by triosephosphate isomerase inhibition by phosphoenolpyruvate, the PKM substrate.[2]
Function
R5P and its derivatives serve as precursors to many biomolecules, including DNA, RNA, ATP, coenzyme A, FAD (Flavin adenine dinucleotide), and histidine.[5]
Nucleotide biosynthesis
Nucleotides serve as the building blocks for nucleic acids, DNA and RNA.[6] They are composed of a nitrogenous base, a pentose sugar, and at least one phosphate group. Nucleotides contain either a purine or a pyrimidine nitrogenous base. All intermediates in purine biosynthesis are constructed on a R5P "scaffold".[7] R5P also serves as an important precursor to pyrimidine ribonucleotide synthesis.
During nucleotide biosynthesis, R5P undergoes activation by ribose-phosphate diphosphokinase (PRPS1) to form phosphoribosyl pyrophosphate (PRPP). Formation of PRPP is essential for both the de novo synthesis of purines and for the purine salvage pathway.[8] The de novo synthesis pathway begins with the activation of R5P to PRPP, which is later catalyzed to become phosphoribosylamine, a nucleotide precursor. During the purine salvage pathway,[9] phosphoribosyltransferases add PRPP to bases.[10]
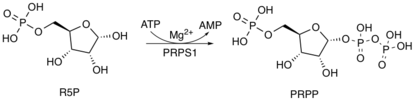
PRPP also plays an important role in pyrimidine ribonucleotide synthesis. During the fifth step of pyrimidine nucleotide synthesis, PRPP covalently links to orotate at the one-position carbon on the ribose unit. The reaction is catalyzed by orotate phosphoriboseyltransferase (PRPP transferase), yielding orotidine monophosphate (OMP).[8]
Histidine biosynthesis
Histidine is an essential amino acid that is not synthesized de novo in humans. Like nucleotides, biosynthesis of histidine is initiated by the conversion of R5P to PRPP. The step of histidine biosynthesis is the condensation of ATP and PRPP by ATP-phosphoribosyl transferase, the rate determining enzyme. Histidine biosynthesis is carefully regulated by feedback inhibition/[11]
Other functions
R5P can be converted to adenosine diphosphate ribose, which binds and activates the TRPM2 ion channel. The reaction is catalyzed by ribose-5-phosphate adenylyltransferase[12]
Disease relevance
Diseases have been linked to R5P imbalances in cells. Cancers and tumors show upregulated production of R5P correlated to increased RNA and DNA synthesis.[2] Ribose 5-phosphate isomerase deficiency, the rarest disease in the world,[13][14] is also linked to an imbalance of R5P. Although the molecular pathology of the disease is poorly understood, hypotheses included decreased RNA synthesis. Another disease linked to R5P is gout[15] Higher levels of G6P lead to a buildup of glycolytic intermediates, that are diverted to R5P production. R5P converts to PRPP, which forces an overproduction of purines, leading to uric acid build up.[8]
Accumulation of PRPP is found in Lesch-Nyhan Syndrome.[16] The build up is caused by a deficiency of the enzyme hypoxanthine-guanine phosphoribosyltransferase (HGPRT), which leads to decreased nucleotide synthesis and an increase of uric acid production.
Superactivity in PRPS1, the enzyme that catalyzes the R5P to PRPP, has also been linked to gout, as well as neurodevelopmental impairment and sensorineural deafness.[17]
References
- Levene PA, Stiller ET (February 1934). "The Synthesis of Ribose-5-Phosphoric Acid". Journal of Biological Chemistry. 104 (2): 299–306.
- Berg JM, Tymoczko JL, Stryer L (2015). Biochemistry (7th ed.). W.H. Freeman. pp. 589–613. ISBN 978-1-4292-7635-1.
- Kruger NJ, von Schaewen A (June 2003). "The oxidative pentose phosphate pathway: structure and organisation". Current Opinion in Plant Biology. 6 (3): 236–46. doi:10.1016/s1369-5266(03)00039-6. PMID 12753973.
- Zhang R, Andersson CE, Savchenko A, Skarina T, Evdokimova E, Beasley S, Arrowsmith CH, Edwards AM, Joachimiak A, Mowbray SL (January 2003). "Structure of Escherichia coli ribose-5-phosphate isomerase: a ubiquitous enzyme of the pentose phosphate pathway and the Calvin cycle". Structure. 11 (1): 31–42. doi:10.1016/s0969-2126(02)00933-4. PMC 2792023. PMID 12517338.
- Coleman JP, Smith CJ (2007). X Pharm: The Comprehensive Pharmacology Reference. pp. 1–6. doi:10.1016/b978-008055232-3.60227-2. ISBN 9780080552323.
- "Nucleotides". IUPAC Compendium of Chemical Terminology. International Union of Pure and Applied Chemistry. 2009. doi:10.1351/goldbook.n04255. ISBN 978-0-9678550-9-7.
- Engelking LR (2015). "Purine Biosynthesis". Textbook of Veterinary Physiological Chemistry (Third ed.). pp. 88–92. doi:10.1016/b978-0-12-391909-0.50015-3. ISBN 978-0-12-391909-0.
- Pelley JW (2011). "Purine, Pyrimidine, and Single-Carbon Metabolism". Elsevier's Integrated Review Biochemistry (2nd ed.). pp. 119–124. doi:10.1016/b978-0-323-07446-9.00014-3. ISBN 9780323074469.
- Engelking LR (2015). "Chapter 31 — Carbohydrate Metabolism in Erythrocytes". Textbook of Veterinary Physiological Chemistry (Third ed.). pp. 190–194. doi:10.1016/b978-0-12-391909-0.50031-1. ISBN 978-0-12-391909-0.
- Schramm VL, Grubmeyer C (2004). Phosphoribosyltransferase Mechanisms and Roles in Nucleic Acid Metabolism. Progress in Nucleic Acid Research and Molecular Biology. 78. pp. 261–304. doi:10.1016/s0079-6603(04)78007-1. ISBN 9780125400787. PMID 15210333.
- Ingle RA (January 2011). "Histidine biosynthesis". The Arabidopsis Book. 9: e0141. doi:10.1199/tab.0141. PMC 3266711. PMID 22303266.
- Evans WR, San Pietro A (January 1966). "Phosphorolysis of adenosine diphosphoribose". Archives of Biochemistry and Biophysics. 113 (1): 236–44. doi:10.1016/0003-9861(66)90178-0. PMID 4287446.
- Wamelink MM, Grüning NM, Jansen EE, Bluemlein K, Lehrach H, Jakobs C, Ralser M (September 2010). "The difference between rare and exceptionally rare: molecular characterization of ribose 5-phosphate isomerase deficiency". Journal of Molecular Medicine. 88 (9): 931–9. doi:10.1007/s00109-010-0634-1. hdl:1871/34686. PMID 20499043.
- Huck JH, Verhoeven NM, Struys EA, Salomons GS, Jakobs C, van der Knaap MS (April 2004). "Ribose-5-phosphate isomerase deficiency: new inborn error in the pentose phosphate pathway associated with a slowly progressive leukoencephalopathy". American Journal of Human Genetics. 74 (4): 745–51. doi:10.1086/383204. PMC 1181951. PMID 14988808.
- Jiménez RT, Puig JG (2012). "Purine Metabolism in the Pathogenesis of Hyperuricemia and Inborn Errors of Purine Metabolism Associated With Disease". Gout & Other Crystal Arthropathies. pp. 36–50. doi:10.1016/b978-1-4377-2864-4.10003-x. ISBN 978-1-4377-2864-4.
- Ichida K, Hosoyamada M, Hosoya T, Endou H (2009). "Primary Metabolic and Renal Hyperuricemia". Genetic Diseases of the Kidney. pp. 651–660. doi:10.1016/b978-0-12-449851-8.00038-3. ISBN 978-0-12-449851-8.
- Singer HS, Mink JW, Gilbert DL, Jankovic J (2010). "Inherited Metabolic Disorders Associated with Extrapyramidal Symptoms". Movement Disorders in Childhood. pp. 164–204. doi:10.1016/B978-0-7506-9852-8.00015-1. ISBN 978-0-7506-9852-8.