Thermal rearrangement of aromatic hydrocarbons
Thermal rearrangements of aromatic hydrocarbons are considered to be unimolecular reactions that directly involve the atoms of an aromatic ring structure and require no other reagent than heat. These reactions can be categorized in two major types: one that involves a complete and permanent skeletal reorganization (isomerization), and one in which the atoms are scrambled but no net change in the aromatic ring occurs (automerization).[1] The general reaction schemes of the two types are illustrated in Figure 1.
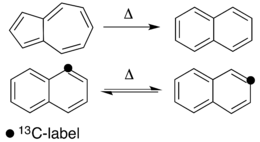
This class of reactions was uncovered through studies on the automerization of naphthalene as well as the isomerization of unsubstituted azulene, to naphthalene. Research on thermal rearrangements of aromatic hydrocarbons has since been expanded to isomerizations and automerizations of benzene and polycyclic aromatic hydrocarbons.
Mechanisms
Automerizations
The first proposed mechanism for a thermal rearrangement of an aromatic compound was for the automerization of naphthalene. It was suggested that the rearrangement of naphthalene occurred due to reversibility of the isomerization of azulene to naphthalene.[2][3] This mechanism would therefore involve an azulene intermediate and is depicted below:
Subsequent work showed that the isomerization of azulene to naphthalene is not readily reversible ( the free energy of a naphthalene to azulene isomerization was too high - approximately 90 kcal/mol).[1] A new reaction mechanism was suggested that involved a carbene intermediate and consecutive 1,2-hydrogen and 1,2-carbon shifts across the same C-C bond but in opposite directions. This is currently the preferred mechanism[4] and is as follows:
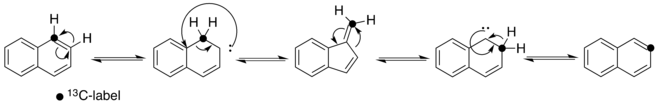
Isomerizations
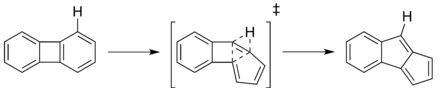
The isomerization of unsubstituted azulene to naphthalene was the first reported thermal transformation of an aromatic hydrocarbon, and has consequently been the most widely studied rearrangement. However, the following mechanisms are generalized to all thermal isomerizations of aromatic hydrocarbons. Many mechanisms have been suggested for this isomerization, yet none have been unequivocally determined as the only correct mechanism. Five mechanisms were originally considered:[1] a reversible ring-closure mechanism, which is shown above, a norcaradiene-vinylidene mechanism, a diradical mechanism, a methylene walk mechanism, and a spiran mechanism. It was quickly determined that the reversible ring-closure mechanism was inaccurate, and it was later decided that there must be multiple reaction pathways occurring simultaneously. This was widely accepted, as at such high temperatures, one mechanism would have to be substantially energetically favored over the others to be occurring alone. Energetic studies displayed similar activation energies for all possible mechanisms.[1]
Four mechanisms for thermal isomerizations have been proposed: a dyotropic mechanism, a diradical mechanism, and two benzene ring contraction mechanisms; a 1,2-carbon shift to a carbene preceding a 1,2-hydrogen shift, and a 1-2-hydrogen shift to a carbene followed by a 1,2-carbon shift.[5][6] The dyotropic mechanism involves concerted 1,2-shifts as displayed below. Electronic studies show this mechanism to be unlikely, but it must still be considered a viable mechanism as it has not yet been disproven.
The diradical mechanism has been supported by kinetic studies performed on the reaction, which have revealed that the reaction is not truly unimolecular, as it is most likely initiated by hydrogen addition from another gas-phase species. However, the reaction still obeys first-order kinetics, which is a classical characteristic of radical chain reactions.[7] A mechanistic rational for the thermal rearrangement of azulene to naphthalene is included below. Homolysis of the weakest bond in azulene occurs, followed by a hydrogen shift and ring closure so as to retain the aromaticity of the molecule.

Benzene ring contractions are the last two mechanisms that have been suggested, and they are currently the preferred mechanisms. These reaction mechanisms proceed through the lowest free energy transition states compared to the diradical and dyotropic mechanisms. The difference between the two ring contractions is minute however, so it has not been determined which is favored over the other. Both mechanisms are shown as follows for the ring contraction of biphenylene:
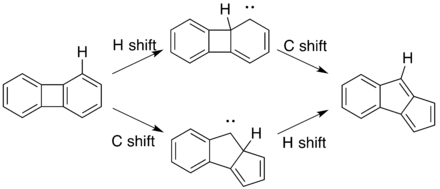
The first involves a 1,2-hydrogen shift to a carbene followed by a 1,2-carbon shift on the same C-C bond but in opposite directions. The second differs from the first only by the order of the 1,2-shifts, with the 1,2-carbon shift preceding the 1,2-hydrogen shift.
The four described mechanisms would all result in the isomerization from azulene to naphthalene. Kinetic data and 13C-labeling have been used to elucidate the correct mechanism, and have led organic chemists to believe that one of the benzene ring contractions is the most likely mechanism through which these isomerizations of aromatic hydrocarbons occur.[5][8]
History
Indications of thermal rearrangements of aromatic hydrocarbons were first noted in the early 20th century by natural products chemists who were working with sesquiterpenes. At the time, they noticed the automerization of a substituted azulene shown below, but no further structural or mechanistic investigations were made.

The oldest characterized thermal rearrangement of an aromatic compound was that of the isomerization of azulene to naphthalene by Heilbronner et al. in 1947.[9] Since then, many other isomerizations have been recorded, however the rearrangement of azulene to naphthalene has received the most attention. Likewise, since the characterization of the automerization of naphthalene by Scott in 1977,[2] similar atom scramblings of other aromatic hydrocarbons such as pyrene,[10] azulene,[3][11] benz[a]anthracene[12] and even benzene have been described.[13] While the existence of these reactions has been confirmed, the isomerization and automerization mechanisms remain unknown.
Reaction conditions and flash vacuum pyrolysis
Thermal rearrangements of aromatic hydrocarbons are generally carried out through flash vacuum pyrolysis (FVP).[14] In a typical FVP apparatus, a sample is sublimed under high vacuum (0.1-1.0 mmHg), heated in the range of 500-1100 °C by an electric furnace as it passes through a horizontal quartz tube, and collected in a cold trap. Sample is carried through the apparatus by nitrogen carrier gas.
FVP has numerous limitations:
- First, it requires a slow rate of sublimation to minimize bimolecular reactions in the gas phase, limiting the amount of material that can be reacted in a given amount of time.
- Second, the high temperatures used in FVP often lead to reactant or product degradation. Combined, these first two limitations restrict FVP yields to the range of 25-30%.[14]
- Third, the high temperatures used in FVP do not allow for the presence of functional groups, thereby limiting possible products.
- Fourth, as FVP is a gas-phase process, difficulties are frequently encountered when scaling above the milligram level.
- Fifth, the FVP synthesis of strained systems mandates temperatures exceeding 1100 °C, which can lead to the degradation and softening of the expensive quartz apparati.[15]
Possible applications
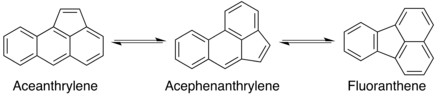
Thermal rearrangements of aromatic hydrocarbons have been shown to be important in areas of chemical research and industry including fullerene synthesis, materials applications, and the formation of soot in combustion.[5] Thermal rearrangements of aceanthrylene and acephenanthrylene can yield fluoranthene, an important species in syntheses of corannulene and fullerenes that proceed through additional internal rearrangements.[8][16]
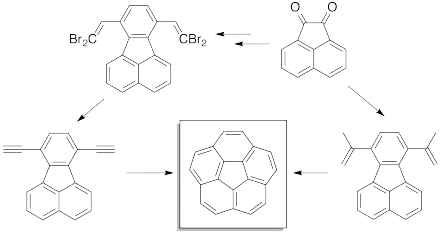
Many of the polycyclic aromatic hydrocarbons known to be tumorigenic or mutagenic are found in atmospheric aerosols, which is connected to the thermal rearrangement of polycyclic aromatic hydrocarbons in fast soot formation during combustion.[16]
References
- Scott, Lawrence T. (1982). "Thermal rearrangements of aromatic compounds". Accounts of Chemical Research. 15 (2): 52–58. doi:10.1021/ar00074a004.
- Scott, Lawrence T.; Agopian, Garabed K. (1977). "Automerization of naphthalene". Journal of the American Chemical Society. 99 (13): 4506–4507. doi:10.1021/ja00455a053.
- Scott, Lawrence T.; Kirms, Mark A. (1981). "Azulene thermal rearrangements. Carbon-13 labeling studies of automerization and isomerization to naphthalene". Journal of the American Chemical Society. 103 (19): 5875–5879. doi:10.1021/ja00409a042.
- Scott, Lawrence T.; Hashemi, Mohammed M.; Schultz, Thomas H.; Wallace, Michael B. (1991). "Thermal rearrangements of aromatic compounds. 15. Automerization of naphthalene. New evidence consistent with the intermediacy of benzofulvene". Journal of the American Chemical Society. 113 (25): 9692–9693. doi:10.1021/ja00025a055.
- Pastor, Michael B.; Kuhn, Ariel J.; Nguyen, Phuong T.; Santander, Mitchell V.; Castro, Claire; Karney, William L. (2013). "Hydrogen shifts and benzene ring contractions in phenylenes". Journal of Physical Organic Chemistry. 26 (9): 750–754. doi:10.1002/poc.3126.
- Cioslowski, Jerzy; Schimeczek, Michael; Piskorz, Pawel; Moncrieff, David (1999). "Thermal Rearrangement of Ethynylarenes to Cyclopentafused Polycyclic Aromatic Hydrocarbons: An Electronic Structure Study". Journal of the American Chemical Society. 121 (15): 3773–3778. doi:10.1021/ja9836601.
- Scott, Lawrence T. (1984). "Thermal rearrangements of aromatic compounds, part 8. Azulene-to-naphthalene rearrangement. A comment on the kinetics". The Journal of Organic Chemistry. 49 (16): 3021–3022. doi:10.1021/jo00190a030.
- Scott, Lawrence T.; Roelofs, Nicolas H. (1987). "Thermal rearrangements of aromatic compounds. 11. Benzene ring contractions at high temperatures. Evidence from the thermal interconversions of aceanthrylene, acephenanthrylene, and fluoranthene". Journal of the American Chemical Society. 109 (18): 5461–5465. doi:10.1021/ja00252a025.
- Heilbronner, E.; Plattner, P. A.; Wieland, K. Rearrangement of Azulene to Naphthalene. Experientia 1947, 3, 70–71.
- Scott, L. T.; Kirms, M. A.; Berg, A.; Hansen, P. E. Automerization of Pyrene a Test for the Mechanism of Naphthalene Automerization. Tetrahedron Letters 1982, 23 (18), 1859–1862. DOI: 10.1016/S0040-4039(00)87204-4
- Becker, Juergen; Wentrup, Curt; Katz, Ellen; Zeller, Klaus Peter (1980). "Azulene-naphthalene rearrangement. Involvement of 1-phenylbuten-3-ynes and 4-phenyl-1,3-butadienylidene". Journal of the American Chemical Society. 102 (15): 5110–5112. doi:10.1021/ja00535a056.
- Scott, L. T.; Tsang, T.-H.; Levy, L. A. Automerizations in Benzenoid Hydrocarbons. New Mechanistic Insights from the Thermal Rearrangement of Benz[a]anthracene-5-13C. Tetrahedron Letters 1984, 25 (16), 1661–1664. DOI: 10.1016/S0040-4039(01)81138-2
- Scott, Lawrence T.; Roelofs, Nicolas H.; Tsang, Tsze Hong (1987). "Thermal rearrangements of aromatic compounds. 10. Automerization of benzene". Journal of the American Chemical Society. 109 (18): 5456–5461. doi:10.1021/ja00252a024.
- Tsefrikas, Vikki M.; Scott, Lawrence T. (2006). "Geodesic Polyarenes by Flash Vacuum Pyrolysis". Chemical Reviews. 106 (12): 4868–4884. doi:10.1021/cr050553y. PMID 17165678.
- Sygula, Andrzej; Rabideau, Peter W. (2006). "Synthesis and Chemistry of Polycyclic Aromatic Hydrocarbons with Curved Surfaces: Buckybowls". Carbon-Rich Compounds. pp. 529–565. doi:10.1002/3527607994.ch12. ISBN 9783527607990.
- Richter, H.; Grieco, W. J.; Howard, J. B. Formation Mechanism of Polycyclic Aromatic Hydrocarbons and Fullerenes in Premixed Benzene Flames. Combustion and Flame 1999, 119 (1–2), 1–22. DOI: 10.1016/S0010-2180(99)00032-2