Ablepharon macrostomia syndrome
Ablepharon macrostomia syndrome (AMS) is an extremely rare, autosomal dominant genetic disorder characterized by abnormal phenotypic appearances that primarily affect the head and face as well as the skull, skin, fingers and genitals. AMS generally results in abnormal ectoderm-derived structures.[1] The most prominent abnormality is the underdevelopment (microblepharon) or absence of eyelids – signifying the ablepharon aspect of the disease – and a wide, fish-like mouth – macrostomia. Infants presenting with AMS may also have malformations of the abdominal wall and nipples. Children with AMS might also experience issues with learning development, language difficulties and intellectual disabilities.
| Ablepharon macrostomia syndrome | |
|---|---|
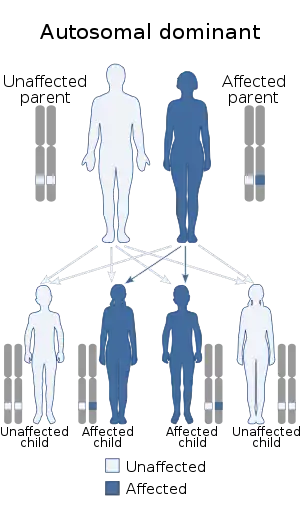 | |
| Ablepharon macrostomia syndrome has an autosomal dominant mode of inheritance. |
AMS is caused by mutations in the TWIST2 gene, among others. It is closely related to Barber–Say syndrome in terms of phenotypic abnormalities.
Signs and symptoms
AMS is generally characterized by abnormal appearances of the skin, eyes, fingers, genitals, head and face. Infants with AMS will have thin, redundantly wrinkled skin and excessive facial creases;[2] wide-set eyes with absent or severely underdeveloped eyelids and down-turned lower eyelids; and a wide, fish-like mouth that may be fused together at the corners. Other appearances of the face and head include: broad nasal bridge, wide, flared nostrils and thick and flared alae nasi (edges of the nostrils).[2]
Abnormalities can also be seen in the hands and fingers, as infants with AMS will also have webbed fingers with limited ability to flex and extend the fingers.[3] Infants with AMS will also display small, rudimentary ears that are atypically low-set on the skull. Absence of the zygomatic bone is also possible. Skin may be dry and coarse, excessively wrinkled around the face and loose around the hands yet tight around the finger joints, leading to diminished use of the fingers.[1][3]
Genital defects may include: ambiguous genitalia, a displaced and/or atypically small penis (micropenis), an absent scrotum around the testes and undescended testicles. Finally, alopecia and thin, sparse hair are also frequently observed.
Causes
Like Barber–Say syndrome, AMS is caused by mutations in the TWIST2 gene that affect a highly conserved residue of TWIST2 (twist-related protein 2). TWIST2 is a basic helix-loop-helix transcription factor that binds to E-box DNA motifs (5'-CANNTG-3') as a heterodimer and inhibits transcriptional activation.[4] Because TWIST2 mediates mesenchymal stem cell differentiation[5] and prevents premature or ectopic osteoblast differentiation,[6] mutations in TWIST2 that disrupt these functions by altering DNA-binding activity could explain many of the phenotypes of AMS.[7] Current research points to the substitution of the wild-type amino acid for Lysine at TWIST2 residue 75 as a significant genetic cause of AMS.[7]
AMS is inherited in an autosomal dominant manner, in which an affected individual needs only one copy of the mutant allele in order to express the disease.[7][8]
Mechanism
The mesenchyme is a mesodermal embryonic tissue that can develop into a multitude of different tissues depending on the needs of the developing embryo. The mesenchyme can develop into blood, cartilage, and membranes. In a normal patient, TWIST2 is highly expressed during embryonic development, specifically in the craniofacial development and chondrogenisis. TWIST2 works to prevent the premature maturation of chondrogenic cells and osteoblasts, the cells that will form cartilage and bone respectively. The dominant mutation in TWIST2 leads to the chondrogenic and osteoblastic cells becoming mature prematurely. This then leads to the primary craniofacial deformities seen in AMS patients.[7]
Diagnosis
Ablepharon macrostomia syndrome can be diagnosed at birth by identification of characteristic physical findings, clinical evaluation, and specialized imaging techniques such as CT scans.[9] CT scans can confirm the absence of the zygomatic arch and abnormalities in the cranial and mandibular bones. An ophthalmologist can diagnose abnormalities in the eyelids and confirm microblepharon or ablepharon. Teams of specialist will typically work together to confirm diagnosis and assess treatment options. Pediatricians, gastroenterologists, dermatologists, urologists, and other care providers can be expected to aid in the diagnosis and treatment.
Treatment
Primary treatment focuses on relief of immediate symptoms such as providing lubrication to the eyes to relieve pain and dryness; antibiotics may also be prescribed to prevent infections and inflammation. Surgical measures can be taken and a plastic surgeon can correct the lack of eyelids through reconstructive surgery.[9] Surgery to correct malformations of the mouth, ears, genitals, fingers, and skin can also be performed as necessary. Macrostomia, the wide, fish-like mouth, can be corrected by a maxillofacial surgeon. The skin can be treated by means of creams to alleviate dryness and coarseness; in certain cases, botulinum toxin and skin grafts were used to improve the overall appearance. It is highly recommended that patients are able to seek the help of pediatric psychologists throughout the entire treatment process.[10]
Prognosis
While there is no cure for AMS, treatment plans provided by doctors can help improve development,[11] overall quality of life, and physical appearance. Physical appearance cannot be corrected to the "norm" but the life expectancy of patients diagnosed with AMS is normal.[12]
Research
Current research into AMS focuses on both underlying causes of the disease and surgical methods for treatment. Currently, a study in Tokyo, Japan is focusing on the role of other TWIST genes in AMS development, specifically the role of TWIST1 and the amino acid substitution that must occur to mutate the gene. TWIST1 mutations are believed to lead to craniosynostosis and ablepharon.[13]
Clinical research focuses on the different surgical techniques used to treat the ablepharon aspect of AMS. The primary goal of such research is to determine which methods are most effective for the patient without being unnecessarily complex. According to a study conducted by the departments of ophthalmology in Sao Paulo and Lima, Peru, full thickness skin grafts have been shown to effectively treat microblepharon in patients with AMS without needing complicated surgeries.[14]
References
- Ciriaco P, Carretta A, Negri G (August 2019). "Laryngo-tracheal stenosis in a woman with ablepharon macrostomia syndrome". BMC Pulmonary Medicine. 19 (1): 163. doi:10.1186/s12890-019-0921-8. PMC 6712709. PMID 31462237.
- De Maria B, Mazzanti L, Roche N, Hennekam RC (August 2016). "Barber-Say syndrome and Ablepharon-Macrostomia syndrome: An overview". American Journal of Medical Genetics. Part A. 170 (8): 1989–2001. doi:10.1002/ajmg.a.37757. PMID 27196381.
- "Ablepharon-Macrostomia Syndrome". NORD (National Organization for Rare Disorders). Retrieved 2019-11-07.
- "TWIST2 - Twist-related protein 2 - Homo sapiens (Human) - TWIST2 gene & protein". www.uniprot.org. Retrieved 2019-01-25.
- Isenmann S, Arthur A, Zannettino AC, Turner JL, Shi S, Glackin CA, Gronthos S (October 2009). "TWIST family of basic helix-loop-helix transcription factors mediate human mesenchymal stem cell growth and commitment". Stem Cells. 27 (10): 2457–68. doi:10.1002/stem.181. PMID 19609939.
- Lee MS, Lowe G, Flanagan S, Kuchler K, Glackin CA (November 2000). "Human Dermo-1 has attributes similar to twist in early bone development". Bone. 27 (5): 591–602. doi:10.1016/S8756-3282(00)00380-X. PMID 11062344.
- Marchegiani S, Davis T, Tessadori F, van Haaften G, Brancati F, Hoischen A, et al. (July 2015). "Recurrent Mutations in the Basic Domain of TWIST2 Cause Ablepharon Macrostomia and Barber-Say Syndromes". American Journal of Human Genetics. 97 (1): 99–110. doi:10.1016/j.ajhg.2015.05.017. PMC 4572501. PMID 26119818.
- Rohena L, Kuehn D, Marchegiani S, Higginson JD (April 2011). "Evidence for autosomal dominant inheritance of ablepharon-macrostomia syndrome". American Journal of Medical Genetics. Part A. 155A (4): 850–4. doi:10.1002/ajmg.a.33900. PMID 21595001.
- "Ablepharon-Macrostomia Syndrome". NORD (National Organization for Rare Disorders). Retrieved 2019-04-19.
- Larumbe J, Villalta P, Velez I (2011). "Clinical variant of ablepharon macrostomia syndrome". Case Reports in Dermatological Medicine. 2011: 593045. doi:10.1155/2011/593045. PMC 3504267. PMID 23198177.
- "Ablepharon macrostomia syndrome". Global Genes. Retrieved 2019-12-13.
- "Ablepharon-Macrostomia Syndrome | Hereditary Ocular Diseases". disorders.eyes.arizona.edu. Retrieved 2019-12-13.
- Takenouchi T, Sakamoto Y, Sato H, Suzuki H, Uehara T, Ohsone Y, Kosaki K (December 2018). "Ablepharon and craniosynostosis in a patient with a localized TWIST1 basic domain substitution". American Journal of Medical Genetics. Part A. 176 (12): 2777–2780. doi:10.1002/ajmg.a.40525. PMID 30450715.
- Cruz AA, Quiroz D, Boza T, Wambier SP, Akaishi PS (2020). "Long-Term Results of the Surgical Management of the Upper Eyelids in "Ablepharon"-Macrostomia Syndrome". Ophthalmic Plastic and Reconstructive Surgery. 36 (1): 21–25. doi:10.1097/IOP.0000000000001442. PMID 31373987.
External links
| Classification |
|---|