Timothy syndrome
Timothy syndrome is a rare autosomal-dominant disorder characterized by physical malformations, as well as neurological and developmental defects, including heart QT-prolongation, heart arrhythmias, structural heart defects, syndactyly (webbing of fingers and toes), and autism spectrum disorders. Timothy syndrome often ends in early childhood death.
| Timothy syndrome | |
|---|---|
| Specialty | Neurology |
Signs and symptoms
The most striking sign of Timothy syndrome is the co-occurrence of both syndactyly (about 0.03% of births) and long QT syndrome (1% per year) in a single patient. Other common symptoms include cardiac arrhythmia (94%), heart malformations (59%), and autism or an autism spectrum disorder (80% who survive long enough for evaluation). Facial dysmorphologies such as flattened noses also occur in about half of patients. Children with this disorder have small teeth, which due to poor enamel coating, are prone to dental cavities and often require removal. The average age of death due to complications of these symptoms is 2.5 years.[1][2][3]
Atypical Timothy syndrome has largely the same symptoms as the classical form. Differences in the atypical form are the lack of syndactyly, the presence of musculoskeletal problems (particularly hyperflexible joints), and atrial fibrillation. Patients with atypical Timothy syndrome also have more facial deformities, including protruding foreheads and tongues. Finally, one patient with atypical Timothy syndrome had a body development discrepancy wherein her upper body was normally developed (that of a 6-year-old) while her lower half resembled a 2- or 3-year-old.[4]
Children with Timothy syndrome tend to be born via caesarean section due to fetal distress.[1][2]
Pathophysiology
There are two recognized types of Timothy syndrome, classical (type-1) and atypical (type-2). They are both caused by mutations in CACNA1C, the gene encoding the calcium channel Cav1.2 α subunit. Timothy syndrome mutations in CACNA1C cause delayed channel closing, thus increased cellular excitability.
Both classical and atypical Timothy syndromes are caused by mutations in CACNA1C. These mutations are in exon 8 (atypical form) and exon 8a (classical form), an alternatively spliced exon. Exon 8a is highly expressed in the heart, brain, gastrointestinal system, lungs, immune system, and smooth muscle. Exon 8 is also expressed in these regions and its level is roughly five-fold higher than exon 8a expression.
One mutation is found in patients with classical Timothy syndrome, G406R, located just past the sixth membrane-spanning segment of domain 1 (D1S6). The conserved glycine at this position seems to be vital for proper voltage-dependent inactivation, as the mutant is lacking in this respect.[3] Atypical Timothy syndrome mutations are similar, one being the identical G406R mutation in the other splice form and the second mutation being G402S, located a few amino acids upstream. The effect of these mutations on channel function is identical to the G406R mutation in classical Timothy syndrome.[4] The lack of proper voltage-dependent inactivation in these mutants causes prolonged inward current and depolarization during cardiac action potentials. This leads to long QT syndrome and resultant arrhythmia. Because exon 8 has greater expression in the heart versus exon 8a, patients with atypical Timothy syndrome have worsened cardiac defects compared to those with the classical form.
Diagnosis
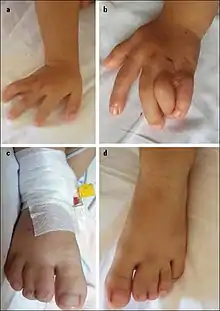
Syndactyly and other deformities are typically observed and diagnosed at birth. Long QT syndrome sometimes presents itself as a complication due to surgery to correct syndactyly. Other times, children collapse spontaneously while playing. In all cases, it is confirmed with ECG measurements. Sequencing of the CACNA1C gene further confirms the diagnosis.
Treatment
Surgery is typically used to correct structural heart defects and syndactyly. Propanolol or beta-adrenergic blockers are often prescribed, as well as insertion of a pacemaker to maintain proper heart rhythm. With the characterization of Timothy syndrome mutations indicating that they cause defects in calcium currents, calcium channel blockers may be effective as a therapeutic agent.[4]
Prognosis
The prognosis for patients diagnosed with Timothy syndrome is very poor. Of 17 children analyzed in one study, 10 died at an average age of 2.5 years. Of those that did survive, three were diagnosed with autism, one with an autism spectrum disorder, and the last had severe delays in language development.[3] One patient with atypical Timothy syndrome was largely normal with the exception of heart arrhythmia.[4] Likewise, the mother of two Timothy syndrome patients also carried the mutation, but lacked any obvious phenotype. In both of these cases, however, the lack of severity of the disorder was due to mosaicism.
History
Some of the abnormalities observed in Timothy syndrome were described in the 1990s. However, it was linked with calcium channel abnormalities in 2004, and the disorder was thence named "Timothy syndrome" in honor of Katherine W. Timothy, who was among the first to identify a case and performed much of the phenotypic analysis that revealed other abnormalities.[3]
References
- Marks M, Whisler S, Clericuzio C, Keating M (1995). "A new form of long QT syndrome associated with syndactyly". J Am Coll Cardiol. 25 (1): 59–64. doi:10.1016/0735-1097(94)00318-K. PMID 7798527.
- Marks M, Trippel D, Keating M (1995). "Long QT syndrome associated with syndactyly identified in females". Am J Cardiol. 76 (10): 744–745. doi:10.1016/S0002-9149(99)80216-1. PMID 7572644.
- Splawski I, Timothy K, Sharpe L, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz P, Joseph R, Condouris K, Tager-Flusberg H, Priori S, Sanguinetti M, Keating M (2004). "Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism". Cell. 119 (1): 19–31. doi:10.1016/j.cell.2004.09.011. PMID 15454078.
- Splawski I, Timothy K, Decher N, Kumar P, Sachse F, Beggs A, Sanguinetti M, Keating M (2005). "Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations". Proc Natl Acad Sci USA. 102 (23): 8089–8096. Bibcode:2005PNAS..102.8089S. doi:10.1073/pnas.0502506102. PMC 1149428. PMID 15863612.
External links
| Classification |
|---|