Cornelia de Lange syndrome
Cornelia de Lange syndrome (CdLS) is a genetic disorder. People with this syndrome experience a range of physical, cognitive, and medical challenges ranging from mild to severe. The syndrome has a widely varied phenotype, meaning people with the syndrome have varied features and challenges. The typical features of CdLS include thick or long eyebrows, a small nose, small stature, developmental delay, long or smooth philtrum, thin upper lip and downturned mouth.[1]
| Cornelia de Lange syndrome | |
|---|---|
| Other names | Bushy syndrome |
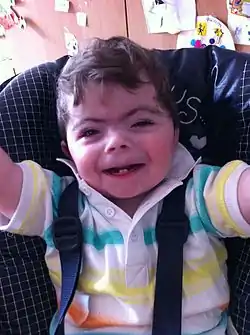 | |
| One-year-old boy with Cornelia de Lange syndrome | |
| Specialty | Medical genetics |
The syndrome is named after Dutch pediatrician Cornelia Catharina de Lange, who described it in 1933.
It is often termed Brachmann de Lange syndrome or Bushy syndrome and is also known as Amsterdam dwarfism. Its exact incidence is unknown, but it is estimated at 1 in 10,000 to 30,000.
Signs and symptoms
The phenotype of CdLS is highly varied and is described as a spectrum; from Classic CdLS (with a greater number of key features) to mild variations with only a few features. Some people will have a small number of features but don't have CdLS.[1]
Key features:
- Long and/or thick eyebrows
- Short nose
- Concave nasal ridge and/or upturned nasal tip
- Long and/or smooth philtrum
- Thin upper lip vermilion and/or downturned corners of mouth
- Missing fingers or toes
- Congenital diaphragmatic hernia
Other suggestive features:
- Developmental delay and/or intellectual disability
- Small prenatal and birth size / weight
- Small stature
- Microcephaly (prenatally and/or postnatally)
- Small hands and/or feet
- Short fifth finger
- Hirsutism
The following health conditions are more common in people with CdLS than in the general population.
- Respiratory illness
- Heart defects (e.g., pulmonary stenosis, VSD, ASD, coarctation of the aorta)
- Hearing impairment
- Vision abnormalities (e.g., ptosis, nystagmus, high myopia, hypertropia)
- Partial joining of the second and third toes
- Incurved 5th fingers (clinodactyly)
- Gastroesophageal reflux
- Gastrointestinal abnormalities
- Musculoskeletal problems
- Scoliosis
- Social anxiety
- Seizures
- Cleft palate
- Feeding problems
Children with this syndrome are often found to have long eyelashes, bushy eyebrows and synophrys (joined eyebrows). Body hair can be excessive and affected individuals are often shorter than their immediate family members. They present a characteristic facial phenotype.[2]
Children with CdLS often suffer from gastrointestinal tract difficulties, particularly gastroesophageal reflux. Vomiting, intermittent poor appetite, constipation, diarrhea or gaseous distention are known to be a regularity in cases where the GI tract problems are acute. Symptoms may range from mild to severe.
People with CdLS may exhibit behaviours that have been described as "autistic-like," including self-stimulation, aggression, self-injury or strong preference to a structured routine. Behavior problems in CdLS are not inevitable. Many behaviour issues associated with CdLS are reactive (i.e., something happens within the person's body or environment to bring on the behavior) and cyclical (comes and goes). Often, an underlying medical issue, pain, social anxiety, environmental or caregiver stress can be associated with a change behaviour. If pain or a medical issue is the cause, once treated, the behaviour diminishes.
There is evidence for some features of premature aging including the early development of Barrett’s esophagus, osteoporosis present in the teenage years, premature greying of hair and some changes to the skin of the face causing a more aged appearance compared to chronological age.[3]
Causes
The vast majority of cases are thought to be due to spontaneous genetic mutations.[1] It can be associated with mutations affecting the cohesin complex.[4]
As of 2018, it was confirmed that 500 genetic mutations have been associated with the condition; occurring on 7 different genes. In around 30% of cases of CdLS the genetic cause remains undiscovered. The wide variation in phenotype is attributed to a high degree of somatic mosaicism in CdLS as well as the different genes and type of mutations. For this reason people with CdLS can have very different appearance, abilities, and associated health issues.[5]
| Name | OMIM | Gene | Appx. % | Notes |
|---|---|---|---|---|
| CDLS1 | 122470 | NIPBL | 50% | A gene responsible for CdLS on chromosome 5 was discovered in 2004 jointly by researchers at the Children's Hospital of Philadelphia, USA[6] and researchers at Newcastle University, UK.[7] |
| CDLS2 | 300590 | SMC1A | 5% | In 2006, a second gene, on the X chromosome, was found by Italian scientists. |
| CDLS3 | 610759 | SMC3 | 1% | A third gene discovery was announced in 2007. The gene is on chromosome 10 and was also discovered by the research team in Philadelphia. |
The latter two genes seem to correlate with a milder form of the syndrome.
In 2004, researchers at the Children's Hospital of Philadelphia (United States) and the University of Newcastle upon Tyne (England) identified a gene (NIPBL) on chromosome 5 that causes CdLS when it is mutated. Since then, additional genes have been found (SMC1A, SMC3 and HDAC8, RAD21) that cause CdLS when changed. In July 2012, the fourth "CdLS gene"—HDAC8—was announced. HDAC8 is an X-linked gene, meaning it is located on the X chromosome. Individuals with CdLS who have the gene change in HDAC8 make up just a small portion of all people with CdLS.[8] Evidence of a linkage at chromosome 3q26.3 is mixed.[9]
Genetic alterations associated with CdLS have been identified in genes NIPBL, SMC1A and SMC3 as well as the more recently identified genes RAD21 and HDAC8.[10] All of these genetic alterations occurring in CdLS patients affect proteins that function in the cohesin pathway.[10] SMC1A, SMC3 and RAD21 proteins are structural components of the cohesin ring complex. NIPBL is involved in the loading of the cohesin ring onto chromosomes, and HDAC8 deacylates SMC3 to facilitate its function. The cohesin pathway is involved in cohesion of sister chromatids during mitosis, DNA repair, chromosome segregation and the regulation of developmental gene expression. Defects in these functions are theorised to underlie some of the features of CdLS.[11] In particular, defective DNA repair may underlie the features of premature aging.[3]
Diagnosis
The diagnosis of CdLS is primarily based on clinical findings by a clinical geneticist; and in some cases may be confirmed through laboratory testing.[5]
Treatment
Often, an interdisciplinary approach is recommended to treat the issues associated with CdLS. A team for promoting the child's well-being often includes speech, occupational and physical therapists, teachers, physicians, and parents.[12]
In August 2020 Briton Major Chris Brannigan walked the length of Britain barefoot to raise half a million pounds, which is being used to engineer the first gene-targeted therapy for Cornelia de Lange Syndrome. If successful this will be the first treatment for CdlS. [13]
History
The first documented case was in 1916 by Winfried Robert Clemens Brachmann (1888–1969), a German physician who wrote about the distinct features of the disease from his 19-year-old patient,[14] followed in 1933 by Cornelia Catharina de Lange (1871–1950),[15] a Dutch pediatrician after whom the disorder has been named.[16] CdLS was formerly known as Brachmann-de Lange Syndrome.[17]
References
- Hennekam, Raoul C.; Balkom, Ingrid D. C. Van; Tümer, Zeynep; Shi, Angell; Rigamonti, Claudia; Richman, David; Redeker, Egbert; Quaglio, Ana L.; Potter, Carol J. (October 2018). "Diagnosis and management of Cornelia de Lange syndrome: first international consensus statement" (PDF). Nature Reviews Genetics. 19 (10): 649–666. doi:10.1038/s41576-018-0031-0. ISSN 1471-0064. PMID 29995837.
- Basel-Vanagaite, L.; Wolf, L.; Orin, M.; Larizza, L.; Gervasini, C.; Krantz, I.D.; Deardoff, M.A. (2016). "Recognition of the Cornelia de Lange syndrome phenotype with facial dysmorphology novel analysis". Clinical Genetics. 89 (5): 557–563. doi:10.1111/cge.12716. PMID 26663098.
- Kline AD, Grados M, Sponseller P, Levy HP, Blagowidow N, Schoedel C, Rampolla J, Clemens DK, Krantz I, Kimball A, Pichard C, Tuchman D (2007). "Natural history of aging in Cornelia de Lange syndrome". Am J Med Genet C Semin Med Genet. 145C (3): 248–60. doi:10.1002/ajmg.c.30137. PMC 4902018. PMID 17640042.
- Liu J, Krantz ID (October 2009). "Bushy Syndrome, cohesin, and beyond". Clin. Genet. 76 (4): 303–14. doi:10.1111/j.1399-0004.2009.01271.x. ISSN 0009-9163. PMC 2853897. PMID 19793304.
- Kline, Antonie D.; Moss, Joanna F.; Selicorni, Angelo; Bisgaard, Anne-Marie; Deardorff, Matthew A.; Gillett, Peter M.; Ishman, Stacey L.; Kerr, Lynne M.; Levin, Alex V. (2018-07-11). "Diagnosis and management of Cornelia de Lange syndrome: first international consensus statement" (PDF). Nature Reviews Genetics. 19 (10): 649–666. doi:10.1038/s41576-018-0031-0. ISSN 1471-0056. PMID 29995837.
- Krantz ID, McCallum J, DeScipio C, et al. (2004). "Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B". Nature Genetics. 36 (6): 631–5. doi:10.1038/ng1364. PMC 4902017. PMID 15146186.
- Tonkin E, Wang TJ, Lisgo S, Bamshad MJ, Strachan T (2004). "NIPBL, encoding a homolog of fungal Scc2-type sister chromatid cohesion proteins and fly Nipped-B, is mutated in Cornelia de Lange syndrome". Nature Genetics. 36 (6): 636–641. doi:10.1038/ng1363. PMID 15146185.
- "HDAC8 FAQ Sheet" (PDF). CdLS Foundation Web site. Cornelia de Lange Syndrome Foundation. Archived from the original (PDF) on 3 September 2013. Retrieved 12 February 2013.
- Krantz ID, Tonkin E, Smith M, et al. (June 2001). "Exclusion of linkage to the CDL1 gene region on chromosome 3q26.3 in some familial cases of Cornelia de Lange syndrome". American Journal of Medical Genetics. 101 (2): 120–9. doi:10.1002/1096-8628(20010615)101:2<120::AID-AJMG1319>3.0.CO;2-G. PMC 4896160. PMID 11391654.
- Boyle MI, Jespersgaard C, Brøndum-Nielsen K, Bisgaard AM, Tümer Z (2015). "Cornelia de Lange syndrome". Clin. Genet. 88 (1): 1–12. doi:10.1111/cge.12499. PMID 25209348.
- Pié J, Gil-Rodríguez MC, Ciero M, López-Viñas E, Ribate MP, Arnedo M, Deardorff MA, Puisac B, Legarreta J, de Karam JC, Rubio E, Bueno I, Baldellou A, Calvo MT, Casals N, Olivares JL, Losada A, Hegardt FG, Krantz ID, Gómez-Puertas P, Ramos FJ (2010). "Mutations and variants in the cohesion factor genes NIPBL, SMC1A, and SMC3 in a cohort of 30 unrelated patients with Cornelia de Lange syndrome". Am. J. Med. Genet. A. 152A (4): 924–9. doi:10.1002/ajmg.a.33348. PMC 2923429. PMID 20358602.
- "CdLS Foundation – Treatment Protocols". 12 February 2013. Retrieved 12 February 2013.
- https://www.bbc.com/news/amp/uk-england-wiltshire-53757274...
- Brachmann W (1916). "Ein Fall von symmetrischer Monodaktylie durch Ulnadefekt, mit symmetrischer Flughautbildung in den Ellenbeugen, sowie anderen Abnormitaeten (Zwerghaftigkeit, Halsrippen, Behaarung) (A case of symmetrical monodactyly, representing ulnar deficiency, with symmetrical antecubital webbing and other abnormalities, (dwarfism, cervical ribs, hirsutism))". Jahrbuch für Kinderheilkunde und physische Erziehung. 84: 225–235.
- de Lange C (1933). "Sur un type nouveau de degenerescence (typus Amstelodamensis)". Arch. Med. Enfants. 36: 713–719.
- "Brachmann-de Lange syndrome".
- Aitken, Kenneth J. (2009). A-Z of Genetic Factors in Autism: A Handbook for Professionals. London: Jessica Kingsley. pp. 172–173. ISBN 9781843109761. Retrieved December 10, 2015.
External links
| Classification | |
|---|---|
| External resources |
| Wikimedia Commons has media related to Cornelia de Lange syndrome. |
