Laurence–Moon syndrome
Laurence–Moon syndrome (LMS) is a rare autosomal recessive[1] genetic disorder associated with retinitis pigmentosa, spastic paraplegia, and mental disabilities.[2]
| Laurence–Moon syndrome | |
|---|---|
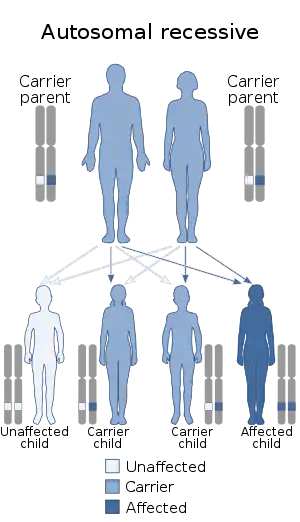 | |
| Laurence–Moon syndrome has an autosomal recessive pattern of inheritance | |
| Specialty | Medical genetics |
Signs and symptoms
Intellectual disability, hexadactyly, central diabetes insipidus, blindness (usually by 30 years due to central retinal degeneration).
Genetics
LMS is inherited in an autosomal recessive manner.[1] This means the defective gene responsible for the disorder is located on an autosome, and two copies of the defective gene (one inherited from each parent) are required in order to be born with the disorder. The parents of an individual with an autosomal recessive disorder both carry one copy of the defective gene, but usually do not experience any signs or symptoms of the disorder.
Diagnosis
The syndrome was originally thought to have five cardinal features (and recently a sixth was added), on the basis of which a diagnostic criteria was developed: 4 primary features or 3 primary features and 2 secondary features must be present.
The primary features are: 1. Polydactyly 2. Rod-cone dystrophy 3. Learning disabilities 4. Obesity 5. Hypogonadism in males 6. Renal abnormalities
While the secondary features are stated to be as: 1. Speech disorder and/or developmental delay 2. Ophthalmic abnormalities other than rod-cone dystrophy (strabismus, cataract, astigmatism etc) 3. Brachydactyly or Syndactyly 4. Polyuria and/or polydipsia (nephrogenic diabetes insipidus) 5. Ataxia, poor coordination, imbalance 6. Mild spasticity (especially lower limbs) 7. Diabetes mellitus 8. Dental crowding, hypodontia, small roots, high arched palate 9. Congenital heart disease 10. Hepatic fibrosis
Treatment
There is no cure to LNMS. However, symptomatic treatment is often provided. The patients with LNMS often experience ataxia, spasticity and contractures, restricting their movements and daily activities. Therefore, multi-disciplinary approach is required including physical therapies, psychiatric and ophthalmogic consultations, nutrition and well-balanced diet. Physical therapy aims at improving the strength and ability using assisting tools such as ankle-foot orthitic braces, weight-bearing walkers and regular exercise.
Eponym and nomenclature
It is named after the physicians John Zachariah Laurence and Robert Charles Moon who provided the first formal description of the condition in a paper published in 1866.[3][4] In the past, LMS has also been referred to as Laurence–Moon–Bardet–Biedl or Laurence–Moon–Biedl–Bardet syndrome, but Bardet–Biedl syndrome (BBS) is now usually recognized as a separate entity.[5]
Recent advances in genetic typing of the phenotypically-wide variation in patients clinically diagnosed with either Bardet-Biedl Syndrome (BBS) or Laurence-Moon Syndrome (LMS) have questioned whether LMS and BBS are genetically distinct. For example, a 1999 epidemiological study of BBS and LMS reported that "BBS proteins interact and are necessary for the development of many organs." "Two patients [in the study] were diagnosed clinically as LMS but both had mutations in a BBS gene. The features in this population do not support the notion that BBS and LMS are distinct."[6] A more recent 2005 paper also suggests that the two conditions are not distinct.[7]
References
- Farag, T. I.; Teebi, A. H. W. S. (Feb 1988). "Bardet-Biedl and Laurence-Moon syndromes in a mixed Arab population". Clinical Genetics. 33 (2): 78–82. doi:10.1111/j.1399-0004.1988.tb03414.x. PMID 3359670. S2CID 43584088.
- "Laurence-Moon Syndrome | Doctor | Patient". Patient. Retrieved 13 December 2016.
- synd/3746 at Who Named It?
- Laurence J.Z., Moon R.C.: Four cases of "retinitis pigmentosa" occurring in the same family, and accompanied by general imperfections of development, Ophthal. Rev. 1866, 2:32–41
- Online Mendelian Inheritance in Man (OMIM): 245800
- Beales P, Elcioglu N, Woolf A, Parker D, Flinter F (1 June 1999). "New criteria for improved diagnosis of Bardet–Biedl syndrome: results of a population survey". J. Med. Genet. 36 (6): 437–46. doi:10.1136/jmg.36.6.437 (inactive 2021-01-10). PMC 1734378. PMID 10874630. Archived from the original on 14 March 2008. Retrieved 21 March 2010.CS1 maint: DOI inactive as of January 2021 (link)
- Moore S, Green J, Fan Y, et al. (2005). "Clinical and genetic epidemiology of Bardet–Biedl syndrome in Newfoundland: a 22-year prospective, population-based, cohort study". Am. J. Med. Genet. A. 132 (4): 352–60. doi:10.1002/ajmg.a.30406. PMC 3295827. PMID 15637713.
External links
| Classification | |
|---|---|
| External resources |