Aminolevulinic acid dehydratase deficiency porphyria
Aminolevulinic acid dehydratase deficiency porphyria (also known as "Doss porphyria",[1] "plumboporphyria",[1] or "ADP"[2]) is a rare autosomal recessive metabolic disorder that results from inappropriately low levels of the enzyme delta-aminolevulinic acid dehydratase (ALAD), which is required for normal heme synthesis. This deficiency results in the accumulation of a toxic metabolic precursor in the heme synthesis pathway called aminolevulinic acid (ALA). [2] Heme is a component of hemoglobin which carries oxygen in red blood cells.
| Aminolevulinic acid dehydratase deficiency porphyria | |
|---|---|
| Other names | Porphyria due to ALA dehydratase deficiency |
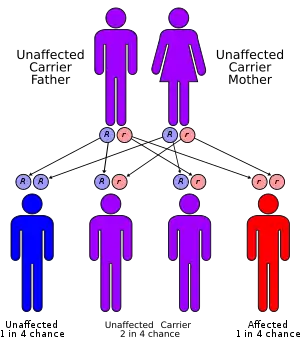 | |
| ALA dehydratase deficiency has an autosomal recessive pattern of inheritance. | |
| Specialty | Gastroenterology, dermatology, medical genetics, endocrinology |
ALA dehydratase deficiency is a rare cause of hepatic porphyria, meaning that excess porphyrins originate from the liver rather than the bone marrow as in erythropoietic porphyrias.[3][4]
Signs and symptoms
The clinical presentation of ADP includes a wide range of neurologic and gastrointestinal symptoms.[2] The severity of symptoms is dependent on how functional the ALAD enzyme is in each person. The higher the enzyme activity, the fewer symptoms a person will exhibit and the later they will present. The less functional a persons ALAD enzyme is, the earlier they will present and the more severe their symptoms will be.
The disease can present during early childhood (as well as in adulthood) with acute neurologic symptoms that resemble those encountered in acute intermittent porphyria.[1] Patients can also have gastrointestinal symptoms during acute attacks, including abdominal cramping, vomiting, and constipation.[2] Gastrointestinal symptoms can result in failure to thrive and poor weight gain in children. Other symptoms that can occur during an acute attack include a rapid heart beat, high blood pressure, and respiratory difficulties.[2]
Acute attacks can last for weeks and are also called "neurovisceral" attacks due to the neurological complications associated. Patients have reported numbness and tingling in the extremities, seizures, burning pain, poor coordination, inability to move muscles voluntarily, and psychological disturbances.[2] Psychosis, though rare, has occurred in severe instances.
Many triggers have been identified for acute ADP attacks including fasting, a low carb diet, dehydration, alcohol intake, the use of estrogen or progesterone, certain drugs, and other mental and physical stressors.[2]
Genetics
ALA dehydratase deficiency is inherited in an autosomal recessive manner.[3] This means a defective gene responsible for the disorder is located on an autosome, and two copies of the defective gene (one inherited from each parent) are required in order to be born with the disorder. The parents of an individual with an autosomal recessive disorder both carry one copy of the defective gene, but usually do not experience any signs or symptoms of the disorder.
In conditions where both parents are carriers:
Diagnosis
To make a diagnosis, the relevant presenting symptoms and a detailed patient history must be considered in addition to obtaining biomarkers in the urine or blood. These biomarkers include urine porphobilinogen (PBG), aminolevulinic acid (ALA), and porphyrins found in blood and urine.[2] PBG levels fluctuate and are best measured during the onset of acute symptoms.[2] ALA levels are increased in ADP and correlate with the severity of the disease.[6]
If levels of porphyrins are significantly elevated, DNA testing can be performed to determine the specific mutations in the ALAD gene. [2][6] DNA analysis is the most specific test for making a diagnosis of ADP.[6]
Treatment
Supportive care and treatment of symptoms are the typical management options for ADP. During acute attacks, patients are often hospitalized and given medications for nausea/vomiting, rapid heartbeat, and hypertension while their fluid and electrolyte levels are monitored.[2] Glucose supplementation and intravenous hematin are the mainstay of treatment for acute attacks.[2] Avoiding physical and psychological stressors has been shown to limit the reoccurrence of attacks.[2][5]
Specific drugs have been identified that can trigger ADP attacks. Inducers of the CYP-450 enzymes are drugs that should be discontinued or avoided in patients with ADP. Drugs included in this category are anti-convulsants like phenytoin and carbamazepine, and other drugs like barbiturates, St. John's wort, and rifampin.
Prevalence
The condition is extremely rare, with fewer than 10 cases ever reported.[7] All reported cases have been seen in males.[2] A feature of ADP that separates it from other porphyrias is that it is more prevalent in males than in females.[2] However, it theoretically affects males and females at the same rate. Most cases have been identified in Europe but it can occur in any population.[5]
See also
- List of cutaneous conditions
References
- Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. ISBN 978-1-4160-2999-1.
- "ALAD-Deficiency Porphyria (ADP)". American Porphyria Foundation. Retrieved 2020-06-29.
- Jaffe EK, Stith L (February 2007). "ALAD porphyria is a conformational disease". American Journal of Human Genetics. 80 (2): 329–37. doi:10.1086/511444. PMC 1785348. PMID 17236137.
- Doss M, von Tiepermann R, Schneider J, Schmid H (October 1979). "New type of hepatic porphyria with porphobilinogen synthase defect and intermittent acute clinical manifestation". Klinische Wochenschrift. 57 (20): 1123–7. doi:10.1007/bf01481493. PMID 513604.
- "ALAD Porphyria". NORD (National Organization for Rare Disorders). Retrieved 2020-06-29.
- "ALA Dehydratase Deficiency Porphyria Workup: Laboratory Studies, Other Tests". emedicine.medscape.com. Retrieved 2020-06-29.
- Overview of the Porphyrias Archived 2011-07-22 at the Wayback Machine at The Porphyrias Consortium (a part of NIH Rare Diseases Clinical Research Network (RDCRN)) Retrieved June 2011