Ataluren
Ataluren, sold under the brand name Translarna, is a medication for the treatment of Duchenne muscular dystrophy. It was designed by PTC Therapeutics.
 | |
 | |
| Clinical data | |
|---|---|
| Trade names | Translarna |
| Other names | PTC124 |
| AHFS/Drugs.com | International Drug Names |
| License data | |
| Routes of administration | By mouth |
| ATC code | |
| Legal status | |
| Legal status | |
| Identifiers | |
| |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.132.097 |
| Chemical and physical data | |
| Formula | C15H9FN2O3 |
| Molar mass | 284.246 g·mol−1 |
| 3D model (JSmol) | |
| |
| |
| | |
Medical use
Ataluren is used in the European Union to treat people with Duchenne muscular dystrophy who have a nonsense mutation in the dystrophin gene, can walk, and are more than five years old.[1]
Contraindications
People who are pregnant or breast feeding should not take ataluren.[1]
Adverse effects
More than 10% of people taking ataluren in clinical trials experienced vomiting; more than 5% experienced diarrhea, nausea, headache, upper abdominal pain, and flatulence; between 1% and 5% of people experienced decreased appetite and weight loss, high levels of triglycerides, high blood pressure, cough, nosebleeds, abdominal discomfort, constipation, rashes, pain in their arms, legs, and chest muscles, blood in their urine, urinary incontinence, and fever.[1]
Interactions
Aminoglycosides should not be given to someone taking ataluren, as they interfere with its mechanism of action. Caution should be used with drugs that induce UGT1A9, or that are substrates of OAT1, OAT3, or OATP1B3.[1]
Pharmacology
While a large number of studies failed to identify the biological target of ataluren,[3][4][5][6][7][8] it was discovered to bind and stabilize firefly luciferase, thus explaining the mechanism by which it created a false positive effect on the read through assay.[9][10]
Ataluren is thought to make ribosomes less sensitive to premature stop codons (an effect referred to as "read-through") by promoting insertion of certain near-cognate tRNA at the site of nonsense codons with no apparent effects on downstream transcription, mRNA processing, stability of the mRNA or the resultant protein, thereby making a functional protein similar to the non-mutated endogenous product.[11] It seems to work particularly well for the stop codon 'UGA'.[4][12]
Studies have demonstrated that ataluren treatment increases expression of full-length dystrophin protein in human and mouse primary muscle cells containing the premature stop codon mutation for Duchenne muscular dystrophy and rescues striated muscle function.[12] Studies in mice with the premature stop codon mutation for cystic fibrosis demonstrated increased CFTR protein production and function.[13] Extending on this work, a mechanistic study with yeast and human cells has elucidated the details of ataluren-mediated nonstandard codon-anticodon base pairings which result in specific amino acid substitutions at specific codon positions in the CFTR protein.[11]
The European Medicines Agency review on the approval of ataluren concluded that "the non-clinical data available were considered sufficient to support the proposed mechanism of action and to alleviate earlier concerns on the selectivity of ataluren for premature stop codons."[14]
Chemistry
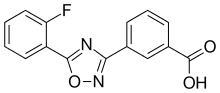
Ataluren is an oxadiazole; its chemical name is 3-[5-(2-Fluorophenyl)-1,2,4-oxadiazol-3-yl]benzoic acid.[4]
History
Ataluren was discovered by scientists at PTC Therapeutics in a collaboration with Lee Sweeney's lab at the University of Pennsylvania, which was initially funded in part by Parent Project Muscular Dystrophy.[15] The team used phenotypic screening of a chemical library to identify compounds that increased the amount of protein expressed by mutated genes, and then optimized one of the hits in the screen to create this drug.[7][5][12] As with the results of many cell-based screens, the biological target of ataluren is not known.[4]
Phase I clinical trials started in 2004.[16]
In 2010, PTC Therapeutics released preliminary results of its phase 2b clinical trial for Duchenne muscular dystrophy, with participants not showing a significant improvement in the six minute walk distance after the 48 weeks of the trial.[17] This failure resulted in the termination of a $100 million deal with Genzyme to pursue the drug.
In May 2014, ataluren received a positive opinion from the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA)[18] and received market authorization from the European Commission to treat people with nonsense mutation Duchenne muscular dystrophy in August 2014;[2] a confirmatory phase III clinical trial was required.[19] By December it was on the market in Germany, France, Italy, Denmark, Spain and a number of other European Union countries.[19]
In February 2016, FDA declined to accept PTC Therapeutics new drug application for ataluren, which was based on a clinical trial in which ataluren missed its primary endpoint; PTC appealed and the FDA declined again in October 2016.[20]
In July 2016, NHS England agreed a Managed Access Agreement (MAA) for Translarna providing reimbursed patient access to Translarna in England via a five-year MAA. This followed a positive recommendation from the National Institute for Health and Care Excellence (NICE) in April 2016, subject to PTC and NHS England finalizing the terms of the MAA. NICE issued its final guidance later in July with implementation of the MAA for patients following within two months.[21]
In March 2017, PTC terminated development of ataluren for cystic fibrosis due to lack of efficacy in the Phase III trials.[22][23][24]
See also
- Biostrophin, rimeporide and eteplirsen, other drugs against Duchenne muscular dystrophy
- Ivacaftor and lumacaftor, other drugs against cystic fibrosis in development by Vertex Pharmaceuticals
References
- "Translarna - Summary of Product Characteristics". UK Electronic Medicines Compendium. 24 April 2017. Retrieved 18 June 2017.
- "Translarna EPAR". European Medicines Agency (EMA). Retrieved 29 September 2020.
- Karijolich, J; Yu, YT (August 2014). "Therapeutic suppression of premature termination codons: mechanisms and clinical considerations (review)". International Journal of Molecular Medicine. 34 (2): 355–62. doi:10.3892/ijmm.2014.1809. PMC 4094583. PMID 24939317.
- Pace, Andrea; Buscemi, Silvestre; Piccionello, Antonio Palumbo; Pibiri, Ivana (2015). "3. Recent Advances in the Chemistry of 1,2,4-Oxadiazoles". In Scriven, Eric F.V.; Ramsden, Christopher A. (eds.). Advances in Heterocyclic Chemistry. Academic Press. p. 127. ISBN 9780128028742.
- Roberts, Roland G. (25 June 2013). "A Read-Through Drug Put through Its Paces". PLOS Biology. 11 (6): e1001458. doi:10.1371/journal.pbio.1001458. PMC 3692443. PMID 23824301.
- Devitt, Liz (25 June 2013). "Researchers question 'read-through' mechanism of muscular dystrophy drug ataluren : Spoonful of Medicine". Nature Medicine: Spoonful of Medicine.
- "Press Release: Questions Raised About Process Used to Identify Experimental Drug for Genetic Disease". NIH via Drug Discovery & Development. 3 February 2009.
- Schmitz, A; Famulok, M (3 May 2007). "Chemical biology: ignore the nonsense". Nature. 447 (7140): 42–3. Bibcode:2007Natur.447...42S. doi:10.1038/nature05715. PMID 17450128. S2CID 29789135.
- Auld, D.S.; Thorne, N.; Maguire, W.F.; Inglese, J. (2009). "Mechanism of PTC124 activity in cell-based luciferase assays of nonsense codon suppression". Proc. Natl. Acad. Sci. USA. 106 (9): 3585–3590. Bibcode:2009PNAS..106.3585A. doi:10.1073/pnas.0813345106. PMC 2638738. PMID 19208811.
- Auld, D.S.; Lovell, S.; Thorne, N.; Lea, W.A.; Maloney, D.J.; v; Rai, G.; Battaile, K.P.; Thomas, C.J.; Simeonov, A.; Hanzlik, R.P.; Inglese, J. (2010). "Molecular basis for the high-affinity binding and stabilization of firefly luciferase by PTC124" (PDF). Proc. Natl. Acad. Sci. USA. 107 (11): 4878–4883. Bibcode:2010PNAS..107.4878A. doi:10.1073/pnas.0909141107. PMC 2841876. PMID 20194791.
- Roy B, Friesen, WJ, Tomizawa Y, Leszyk JD, Zhuo J, Johnson B, Dakka J, Trotta CR, Xue X, Mutyam V, Keeling KM, Mobley JA, Rowe SM, Bedwell DM, Welch EM, Jacobson A (October 2016). "Ataluren stimulates ribosomal selection of near-cognate tRNAs to promote nonsense suppression". Proceedings of the National Academy of Sciences of the United States of America. 113 (44): 12508–12513. doi:10.1073/pnas.1605336113. PMC 5098639. PMID 27702906.
- Welch, EM; Barton, ER; Zhuo, J; Tomizawa, Y; Friesen, WJ; Trifillis, P; Paushkin, S; Patel, M; Trotta, CR; Hwang, S; Wilde, RG; Karp, G; Takasugi, J; Chen, G; Jones, S; Ren, H; Moon, YC; Corson, D; Turpoff, AA; Campbell, JA; Conn, MM; Khan, A; Almstead, NG; Hedrick, J; Mollin, A; Risher, N; Weetall, M; Yeh, S; Branstrom, AA; Colacino, JM; Babiak, J; Ju, WD; Hirawat, S; Northcutt, VJ; Miller, LL; Spatrick, P; He, F; Kawana, M; Feng, H; Jacobson, A; Peltz, SW; Sweeney, HL (3 May 2007). "PTC124 targets genetic disorders caused by nonsense mutations". Nature. 447 (7140): 87–91. Bibcode:2007Natur.447...87W. doi:10.1038/nature05756. PMID 17450125. S2CID 4423529.
- Du M, Liu X, Welch EM, Hirawat S, Peltz SW, Bedwell DM (February 2008). "PTC124 is an orally bioavailable compound that promotes suppression of the human CFTR-G542X nonsense allele in a CF mouse model". Proceedings of the National Academy of Sciences of the United States of America. 105 (6): 2064–9. Bibcode:2008PNAS..105.2064D. doi:10.1073/pnas.0711795105. PMC 2538881. PMID 18272502.
- Haas M, Vlcek V, Balabanov P, Salmonson T, Bakchine S, Markey G, Weise M, Schlosser-Weber G, Brohmann H, Yerro CP, Mendizabal MR, Stoyanova-Beninska V, Hillege HL (January 2015). "European Medicines Agency review of ataluren for the treatment of ambulant patients aged 5 years and older with Duchenne muscular dystrophy resulting from a nonsense mutation in the dystrophin gene". Neuromuscular Disorders. 25 (1): 5–13. doi:10.1016/j.nmd.2014.11.011. PMID 25497400. S2CID 41468577.
- "Press release: PTC Therapeutics Announces $15.4 Million NIH Research Grant for Duchenne Muscular Dystrophy | Evaluate". PTC, University of Pennsylvania, and the NIH via Evaluate Group. July 10, 2007.
- "Press Release: PTC Therapeutics Initiates Clinical Trials of PTC124 for Cystic Fibrosis and Duchenne Muscular Dystrophy". PTC Therapeutics, Inc via P&T Community. July 13, 2004.
- "PTC Therapeutics and Genzyme Corporation announce preliminary results from the phase 2b clinical trial of ataluren for nonsense mutation Duchenne/Becker muscular dystrophy (NASDAQ:PTCT)". Ptct.client.shareholder.com. Archived from the original on 2015-07-04. Retrieved 2013-11-28.
- "PTC Therapeutics Receives Positive Opinion from CHMP for Translarna (ataluren)". MarketWatch.
- "PTC Therapeutics Announces Launch of Translarna (ataluren) in Germany". marketwatch.com. 3 Dec 2014. Retrieved 27 Dec 2014.
- Pagliarulo, Ned (October 17, 2016). "FDA snubs PTC appeal for Duchenne drug". BioPharma Dive.
- "NHS England » NHS England successfully negotiates access to new drug treatment for children with duchenne muscular dystrophy". www.england.nhs.uk.
- "Drug Company Ends Ataluren Program for CF Nonsense Mutations". Cystic Fibrosis Foundation. March 3, 2017.
- DeFrancesco, Laura (9 May 2017). "Drug pipeline: 1Q17". Nature Biotechnology. 35 (5): 400. doi:10.1038/nbt.3874. PMID 28486449. S2CID 205284732.
- Aslam, AA; Higgins, C; Sinha, IP; Southern, KW (19 January 2017). "Ataluren and similar compounds (specific therapies for premature termination codon class I mutations) for cystic fibrosis". The Cochrane Database of Systematic Reviews. 1: CD012040. doi:10.1002/14651858.CD012040.pub2. PMC 6464785. PMID 28102546.
External links
- "Ataluren". Drug Information Portal. U.S. National Library of Medicine.
