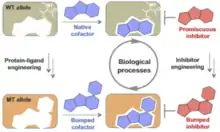
Bump and hole
The bump-and-hole method is a tool in chemical genetics for studying a specific isoform in a protein family without perturbing the other members of the family. The unattainability of isoform-selective inhibition due to structural homology in protein families is a major challenge of chemical genetics. With the bump-and-hole approach, a protein–ligand interface is engineered to achieve selectivity through steric complementarity while maintaining biochemical competence and orthogonality to the wild type pair. Typically, a "bumped" ligand/inhibitor analog is designed to bind a corresponding "hole-modified" protein. Bumped ligands are commonly bulkier derivatives of a cofactor of the target protein. Hole-modified proteins are recombinantly expressed with an amino acid substitution from a larger to smaller residue, e.g. glycine or alanine, at the cofactor binding site. The designed ligand/inhibitor has specificity for the engineered protein due to steric complementarity, but not the native counterpart due to steric interference.[1]
History
Inspiration for the bump-and-hole method was drawn from mutant E. coli strains which carried an A294S mutant version of phenylalanine tRNA synthetase and survived exposure to p-FluoroPhe, a slightly bumped phenylalanine analog which is cytotoxic when incorporated in translation. The A294S mutant strain was able to incorporate Phe, but not the bumped p-FluoroPhe due to steric crowding from the hydroxymethylene of S294.[2] Later work in the labs of Peter G. Schultz and David A. Tirrell showed that a hole-modified A294G phenylalanine tRNA synthetase mutant was able to incorporate the bumped p-FluoroPhe in translation, demonstrating that steric manipulation can successfully broaden substrate scope, even for the highly specific aminoacyl synthetase.[3]
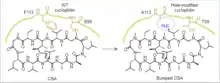
The first bump-and-hole pair, developed by Stuart Schreiber and colleagues, was a bumped cyclosporin A small-molecule with an Ile replacing Val at position 11, and a hole-modified (S99T/F113A) cyclophilin mutant.[5] Cyclosporin A is a chemical inducer of dimerization (CID) of cyclophilin. This first bump-and-hole pair was engineered to improve the binding efficiency between wild type cyclosporin A and cyclophilin, thereby giving more efficient CID. The bumped cyclosporin A was found to interact efficiently with the hole-modified cyclophilin mutant, but not endogenous cyclophilin. The orthogonal CID pair was used to inhibit calcineurin-mediated dephosphorylation of nuclear factor of activated T cells in a cell- and tissue-specific manner.[6] More recently, this first bump-and-hole pair was used to induce the assembly of ten-eleven translocation 2 dioxygenase in cells for temporally controlled DNA demethylation.[7]
Applications
As structural information about protein-ligand interfaces have become available, bump-and-hole pairs have been used to elucidate the substrates of specific proteins from various protein classes, as well as develop orthogonal neoenzyme-neosubstrate therapeutics.
Kinases
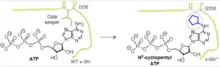
Human protein kinases use ATP as a cofactor to phosphorylate substrate proteins. Kinases play critical roles in complex cell signaling networks. Conserved ATP binding sites and similar catalytic mechanisms pose a challenge to selectively inhibiting a particular kinase to determine its function. Kevan Shokat's lab has developed bump-and-hole pairs using kinase mutants with bulky "gatekeeper" residues in the ATP-binding pocket replaced by Gly or Ala, and bulky ATP analogs. In early work, v-Src kinase I338A/G mutants were shown to accept [γ-32P]-labeled bumped N6-cyclopentyl and N6-benzyl ATP analogs as alternative cofactors to radiolabel its substrates.[8] Only the mutant kinase was able to bind the bumped ATP analogs, allowing labeling of substrates specific to the engineered v-Src kinase. Purification and MS-based proteomics yielded the substrates of v-Src kinase. Hole-modified kinase and bumped ATP analog pairs enabled substrate profiling of several other kinases, including CDK1, Pho85, ERK2, and JNK.[9]
While bumped ATM analogs can help deconvolute kinase substrate profiles, one drawback of this strategy is the cell impermeability of the bumped analogs. To get around this, the Shokat group demonstrated that a bumped ATP analog, kinetin ATP or KTP, could be synthesized endogenously in cells cultured with kinetin. Once synthesized, it can activate a PINK1 kinase mutant, which is otherwise inactive in the absence of the bumped analog. Inactive PINK1 is implicated in Parkinson's disease (PD). In the context of PD, the mutant PINK1-KTP pair represents an orthogonal neoenzyme-neosubstrate therapeutic.[10]
The Shokat group also applied the bump-and-hole approach to develop selective, cell-permeable bumped inhibitors of mutant kinases. For the I338G v-Src kinase, a 4-amino-l-tert-butyl-3-(p-methylphenyl)pyrazolo[3,4-d]pyrimidine (PP1) derivative called p-tButPhe-PP1 was developed for selective inhibition; steric bulk precluded binding to the wild type v-Src kinase. In mammalian cell lines, active v-Src kinase is required for transformation by Rous sarcoma virus. In cell lines expressing I338G v-Src kinase and transfected with RSV, treatment with p-tButPhe-PP1 caused the reversal of transformation, suggesting inhibition of the kinase mutant.[11] Later, the group developed bumped inhibitors 1-naphthyl PP1 (NA-PP1) and 1-methylnaphthyl PP1 (MN-PP1), which inhibited hole-modified yeast kinases with IC50 values in low nanomolar concentrations.[12]
BET proteins
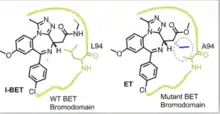
The BET (Bromodomains and Extra Terminal) family of proteins contain conserved motifs known as bromodomains (BDs) responsible for recognizing acetylated lysine on nucleosomal histones.[14] Recently, four members of the BET family, BRD2, 3, 4, and BRDT, each containing two bromodomains, were identified as important regulators of transcription.[15] In order to probe bromodomain-specific functions of members of the BET family, small-molecule inhibitors JQ1 and I-BET were developed, but they lacked inter- and intra-BET (between BDs on the same protein) selectivity.[16] The lab of Alessio Ciulli produced bump-and-hole pairs consisting of ET, a derivative of I-BET with an ethyl bump, and different members of the BET family with an L94A mutation in their BD1.[13] ET was found to have a 160-fold greater specificity for hole-modified BD1 of BET mutants compared to compared to the BDs of wild type BET proteins, giving BD-specific inhibition. The BD-ET bump-and-hole pairs were used to show that selective inhibition of BD1 in a BET protein disrupts chromatin engagement. Recently, the Ciulli group developed a new bump-and-hole pair consisting of BET mutants with a Leu to Val mutation in a BD and the bumped small-molecule inhibitor 9-ME-1. This bumped inhibitor was found to have an IC50 of 200nM and over 100-fold specificity for the L/V BET mutant BD over wild type BDs. This bump-and-hole pair allowed selective inhibition of specific BDs in specific BET proteins, elucidating their role in human cells. It was found that while BD1 is important for chromatin localization of BET proteins, BD2 regulates gene expression by binding and recruiting non-histone acetylated proteins, such as transcription factors.[17]
Glycosidases
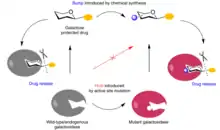
Glycosidases are a family of enzymes that catalyzes the hydrolysis of glycosidic bonds. These enzymes can cleave glycans from glycosylated proteins, one of the most common forms of post-translational modification. In a recent therapeutic application of the bump-and-hole method, a hole-modified galactosidase was paired with a bumped galactosyl-pro-drug. Jingli Hou and colleagues sought to deliver nitric oxide, an important messenger for promoting tissue growth processes like angiogenesis and vasculogenesis, in a spatiotemporally controlled manner. They opted for a pro-drug system, wherein the NO-releasing drug, NONOate, is initially glycosylated. Once the glycosylated NONOate enters cells and is exposed to glycosidases, NO is released. However, non-tissue-specific systemic release of NO, which can reduce therapeutic efficiency and cause harmful side effects, from these pro-drugs was evident due to widespread distribution of endogenous glycosidases. To get around this, Hou et al. developed a bumped pro-drug via methylation of the O6 of the galactose moiety of galactosyl-NONOate. They engineered a corresponding hole-modified β-galactosidase mutant, A4-β-GalH363A with specificity for the bumped galactosyl-NONOate. The bumped pro-drug evaded cleavage by wild type β-galactosidase due to the methylated O6 of the galactose moiety and strict regioselectivity of glycosidases. NO was released in tissues only in the presence of both the bumped galactosyl-NONOate and the hole-modified β-galactosidase mutant, giving spatiotemporal control of delivery. Hou et al. found markedly increased therapeutic efficiency of NO delivery via the bump-and-hole engineered system, compared to the unmodified pro-drug, in rat hindlimb ischemia and mouse acute kidney injury models.[18]
N-Acetylgalactosaminyl transferases
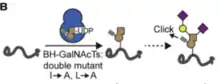
The N-Acetylgalactosaminyl transferase (GalNac Ts) family transfers N-Acetylgalactosamine to the Ser/Thr side chains (O-linked glycosylation) of its substrates, using UDP-GalNac as a cofactor. Like kinases, substrate profiling for specific isoforms of GalNac Ts has been difficult to achieve. The absence of a glycosylation consensus sequence and the variability of glycan elaboration pose a challenge to studying O-GalNac glycoproteins. Further, GalNac transferase knockout strategies are ineffective because the activity of isoforms in the family is both redundant and competitive, such that compensation occurs upon KO. Recently, Schumann et al. applied the bump-and-hole strategy to engineer bumped alkyne-containing UDP-GalNac analogs and double hole-modified I253A/L310A mutant GalNac Ts (BH GalNac Ts). The UDP-alkyne analogs were specific to complementary BH GalNac Ts, which were shown to maintain the biochemical competence of wild type GalNac Ts, with regards to structure, localization, and substrate specificity. This bump-and-hole pair attached a bio-orthogonal label, visualizable through click chemistry, on the substrates of distinct GalNac T isoforms, deconvolving substrate profiles while displaying complexity of glycan elaboration in the secretory pathway.[19]
References
- Islam, Kabirul (October 2018). "The Bump-and-Hole Tactic: Expanding the Scope of Chemical Genetics". Cell Chemical Biology. 25 (10): 1171–1184. doi:10.1016/j.chembiol.2018.07.001. PMC 6195450. PMID 30078633.
- Kast, Peter; Hennecke, Hauke (November 1991). "Amino acid substrate specificity of Escherichia coli phenylalanyl-tRNA synthetase altered by distinct mutations". Journal of Molecular Biology. 222 (1): 99–124. doi:10.1016/0022-2836(91)90740-W. PMID 1942071.
- Liu, Chang C.; Schultz, Peter G. (2010-06-07). "Adding New Chemistries to the Genetic Code". Annual Review of Biochemistry. 79 (1): 413–444. doi:10.1146/annurev.biochem.052308.105824. ISSN 0066-4154. PMID 20307192.
- Belshaw, Peter J.; Schoepfer, Joseph G.; Liu, Karen-Qianye; Morrison, Kim L.; Schreiber, Stuart L. (1995-10-16). "Rational Design of Orthogonal Receptor–Ligand Combinations". Angewandte Chemie International Edition in English. 34 (19): 2129–2132. doi:10.1002/anie.199521291. ISSN 0570-0833.
- Schreiber, Stuart L. (1998-08-01). "Chemical genetics resulting from a passion for synthetic organic chemistry". Bioorganic & Medicinal Chemistry. 6 (8): 1127–1152. doi:10.1016/S0968-0896(98)00126-6. ISSN 0968-0896. PMID 9784856.
- Belshaw, Peter J.; Schreiber, Stuart L. (February 1997). "Cell-Specific Calcineurin Inhibition by a Modified Cyclosporin". Journal of the American Chemical Society. 119 (7): 1805–1806. doi:10.1021/ja9636146. ISSN 0002-7863.
- Lee, Minjung; Li, Jia; Liang, Yi; Ma, Guolin; Zhang, Jixiang; He, Lian; Liu, Yuliang; Li, Qian; Li, Minyong; Sun, Deqiang; Zhou, Yubin (2017-04-05). "Engineered Split-TET2 Enzyme for Inducible Epigenetic Remodeling". Journal of the American Chemical Society. 139 (13): 4659–4662. doi:10.1021/jacs.7b01459. ISSN 0002-7863. PMC 5385525. PMID 28294608.
- Liu, Yi; Shah, Kavita; Yang, Feng; Witucki, Laurie; Shokat, Kevan M. (February 1998). "Engineering Src family protein kinases with unnatural nucleotide specificity". Chemistry & Biology. 5 (2): 91–101. doi:10.1016/S1074-5521(98)90143-0. PMID 9495830.
- Ubersax, Jeffrey A.; Woodbury, Erika L.; Quang, Phuong N.; Paraz, Maria; Blethrow, Justin D.; Shah, Kavita; Shokat, Kevan M.; Morgan, David O. (October 2003). "Targets of the cyclin-dependent kinase Cdk1". Nature. 425 (6960): 859–864. Bibcode:2003Natur.425..859U. doi:10.1038/nature02062. ISSN 0028-0836. PMID 14574415.
- Hertz, Nicholas T.; Berthet, Amandine; Sos, Martin L.; Thorn, Kurt S.; Burlingame, Al L.; Nakamura, Ken; Shokat, Kevan M. (August 2013). "A Neo-Substrate that Amplifies Catalytic Activity of Parkinson's-Disease-Related Kinase PINK1". Cell. 154 (4): 737–747. doi:10.1016/j.cell.2013.07.030. PMC 3950538. PMID 23953109.
- Bishop, Anthony C.; Shah, Kavita; Liu, Yi; Witucki, Laurie; Kung, Chi-yun; Shokat, Kevan M. (February 1998). "Design of allele-specific inhibitors to probe protein kinase signaling". Current Biology. 8 (5): 257–266. doi:10.1016/S0960-9822(98)70198-8. PMID 9501066.
- Bishop, Anthony C.; Ubersax, Jeffrey A.; Petsch, Dejah T.; Matheos, Dina P.; Gray, Nathanael S.; Blethrow, Justin; Shimizu, Eiji; Tsien, Joe Z.; Schultz, Peter G.; Rose, Mark D.; Wood, John L. (September 2000). "A chemical switch for inhibitor-sensitive alleles of any protein kinase". Nature. 407 (6802): 395–401. Bibcode:2000Natur.407..395B. doi:10.1038/35030148. ISSN 0028-0836. PMID 11014197.
- Baud, M. G. J.; Lin-Shiao, E.; Cardote, T.; Tallant, C.; Pschibul, A.; Chan, K.-H.; Zengerle, M.; Garcia, J. R.; Kwan, T. T.- L.; Ferguson, F. M.; Ciulli, A. (2014-10-31). "A bump-and-hole approach to engineer controlled selectivity of BET bromodomain chemical probes". Science. 346 (6209): 638–641. Bibcode:2014Sci...346..638B. doi:10.1126/science.1249830. ISSN 0036-8075. PMC 4458378. PMID 25323695.
- Filippakopoulos, Panagis; Qi, Jun; Picaud, Sarah; Shen, Yao; Smith, William B.; Fedorov, Oleg; Morse, Elizabeth M.; Keates, Tracey; Hickman, Tyler T.; Felletar, Ildiko; Philpott, Martin (December 2010). "Selective inhibition of BET bromodomains". Nature. 468 (7327): 1067–1073. Bibcode:2010Natur.468.1067F. doi:10.1038/nature09504. ISSN 0028-0836. PMC 3010259. PMID 20871596.
- Shi, Jian; Wang, Yifan; Zeng, Lei; Wu, Yadi; Deng, Jiong; Zhang, Qiang; Lin, Yiwei; Li, Junlin; Kang, Tiebang; Tao, Min; Rusinova, Elena (February 2014). "Disrupting the Interaction of BRD4 with Diacetylated Twist Suppresses Tumorigenesis in Basal-like Breast Cancer". Cancer Cell. 25 (2): 210–225. doi:10.1016/j.ccr.2014.01.028. PMC 4004960. PMID 24525235.
- Filippakopoulos, Panagis; Qi, Jun; Picaud, Sarah; Shen, Yao; Smith, William B.; Fedorov, Oleg; Morse, Elizabeth M.; Keates, Tracey; Hickman, Tyler T.; Felletar, Ildiko; Philpott, Martin (December 2010). "Selective inhibition of BET bromodomains". Nature. 468 (7327): 1067–1073. Bibcode:2010Natur.468.1067F. doi:10.1038/nature09504. ISSN 0028-0836. PMC 3010259. PMID 20871596.
- Runcie, A. C.; Zengerle, M.; Chan, K.-H.; Testa, A.; van Beurden, L.; Baud, M. G. J.; Epemolu, O.; Ellis, L. C. J.; Read, K. D.; Coulthard, V.; Brien, A. (2018). "Optimization of a "bump-and-hole" approach to allele-selective BET bromodomain inhibition". Chemical Science. 9 (9): 2452–2468. doi:10.1039/C7SC02536J. ISSN 2041-6520. PMC 5909127. PMID 29732121.
- Hou, Jingli; Pan, Yiwa; Zhu, Dashuai; Fan, Yueyuan; Feng, Guowei; Wei, Yongzhen; Wang, He; Qin, Kang; Zhao, Tiechan; Yang, Qiang; Zhu, Yan (February 2019). "Targeted delivery of nitric oxide via a 'bump-and-hole'-based enzyme–prodrug pair". Nature Chemical Biology. 15 (2): 151–160. doi:10.1038/s41589-018-0190-5. ISSN 1552-4450. PMID 30598545.
- Schumann, Benjamin; Malaker, Stacy Alyse; Wisnovsky, Simon Peter; Debets, Marjoke Froukje; Agbay, Anthony John; Fernandez, Daniel; Wagner, Lauren Jan Sarbo; Lin, Liang; Li, Zhen; Choi, Junwon; Fox, Douglas Michael (April 2020). "Bump-and-Hole Engineering Identifies Specific Substrates of Glycosyltransferases in Living Cells". Molecular Cell: S1097276520301982. doi:10.1016/j.molcel.2020.03.030. PMID 32325029.