Gel electrophoresis of proteins
Protein electrophoresis is a method for analysing the proteins in a fluid or an extract. The electrophoresis may be performed with a small volume of sample in a number of alternative ways with or without a supporting medium: SDS polyacrylamide gel electrophoresis (in short: gel electrophoresis, PAGE, or SDS-electrophoresis), free-flow electrophoresis, electrofocusing, isotachophoresis, affinity electrophoresis, immunoelectrophoresis, counterelectrophoresis, and capillary electrophoresis. Each method has many variations with individual advantages and limitations. Gel electrophoresis is often performed in combination with electroblotting immunoblotting to give additional information about a specific protein. Because of practical limitations, protein electrophoresis is generally not suited as a preparative method.
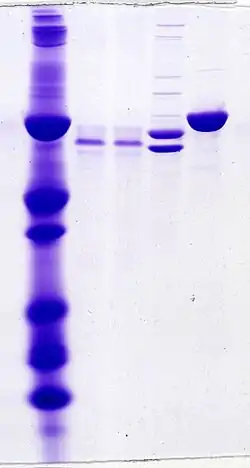
Denaturing gel methods
SDS-PAGE
SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis, describes a collection of related techniques to separate proteins according to their electrophoretic mobility (a function of the molecular weight of a polypeptide chain) while in the denatured (unfolded) state. In most proteins, the binding of SDS to the polypeptide chain imparts an even distribution of charge per unit mass, thereby resulting in a fractionation by approximate size during electrophoresis.
SDS is a strong detergent agent used to denature native proteins to unfolded, individual polypeptides. When a protein mixture is heated to 100 °C in presence of SDS, the detergent wraps around the polypeptide backbone. In this process, the intrinsic charges of polypeptides becomes negligible when compared to the negative charges contributed by SDS. Thus polypeptides after treatment become rod-like structures possessing a uniform charge density, that is same net negative charge per unit length. The electrophoretic mobilities of these proteins will be a linear function of the logarithms of their molecular weights.
Native gel methods
Native gels, also known as non-denaturing gels, analyze proteins that are still in their folded state. Thus, the electrophoretic mobility depends not only on the charge-to-mass ratio, but also on the physical shape and size of the protein.
Blue native PAGE
BN-PAGE is a native PAGE technique, where the Coomassie Brilliant Blue dye provides the necessary charges to the protein complexes for the electrophoretic separation.[1][2] The disadvantage of Coomassie is that in binding to proteins it can act like a detergent causing complexes to dissociate. Another drawback is the potential quenching of chemoluminescence (e.g. in subsequent western blot detection or activity assays) or fluorescence of proteins with prosthetic groups (e.g. heme or chlorophyll) or labelled with fluorescent dyes.
Clear native PAGE
CN-PAGE (commonly referred to as Native PAGE) separates acidic water-soluble and membrane proteins in a polyacrylamide gradient gel. It uses no charged dye so the electrophoretic mobility of proteins in CN-PAGE (in contrast to the charge shift technique BN-PAGE) is related to the intrinsic charge of the proteins.[3] The migration distance depends on the protein charge, its size and the pore size of the gel. In many cases this method has lower resolution than BN-PAGE, but CN-PAGE offers advantages whenever Coomassie dye would interfere with further analytical techniques, for example it has been described as a very efficient microscale separation technique for FRET analyses.[4] Also CN-PAGE is milder than BN-PAGE so it can retain labile supramolecular assemblies of membrane protein complexes that are dissociated under the conditions of BN-PAGE.
Quantitative native PAGE
The folded protein complexes of interest separate cleanly and predictably due to the specific properties of the polyacrylamide gel. The separated proteins are continuously eluted into a physiological eluent and transported to a fraction collector. In four to five PAGE fractions each the metal cofactors can be identified and absolutely quantified by high-resolution ICP-MS. The respective structures of the isolated metalloproteins can be determined by solution NMR spectroscopy.[5]
Buffer systems

Most protein separations are performed using a "discontinuous" (or DISC) buffer system that significantly enhances the sharpness of the bands within the gel. During electrophoresis in a discontinuous gel system, an ion gradient is formed in the early stage of electrophoresis that causes all of the proteins to focus into a single sharp band. The formation of the ion gradient is achieved by choosing a pH value at which the ions of the buffer are only moderately charged compared to the SDS-coated proteins. These conditions provide an environment in which Kohlrausch's reactions determine the molar conductivity. As a result, SDS-coated proteins are concentrated to several fold in a thin zone of the order of 19 μm within a few minutes. At this stage all proteins migrate at the same migration speed by isotachophoresis. This occurs in a region of the gel that has larger pores so that the gel matrix does not retard the migration during the focusing or "stacking" event.[6][7] Separation of the proteins by size is achieved in the lower, "resolving" region of the gel. The resolving gel typically has a much smaller pore size, which leads to a sieving effect that now determines the electrophoretic mobility of the proteins. At the same time, the separating part of the gel also has a pH value in which the buffer ions on average carry a greater charge, causing them to "outrun" the SDS-covered proteins and eliminate the ion gradient and thereby the stacking effect.
A very widespread discontinuous buffer system is the tris-glycine or "Laemmli" system that stacks at a pH of 6.8 and resolves at a pH of ~8.3-9.0. A drawback of this system is that these pH values may promote disulfide bond formation between cysteine residues in the proteins because the pKa of cysteine ranges from 8-9 and because reducing agent present in the loading buffer doesn't co-migrate with the proteins. Recent advances in buffering technology alleviate this problem by resolving the proteins at a pH well below the pKa of cysteine (e.g., bis-tris, pH 6.5) and include reducing agents (e.g. sodium bisulfite) that move into the gel ahead of the proteins to maintain a reducing environment. An additional benefit of using buffers with lower pH values is that the acrylamide gel is more stable at lower pH values, so the gels can be stored for long periods of time before use.[8][9]
SDS gradient gel electrophoresis of proteins
As voltage is applied, the anions (and negatively charged sample molecules) migrate toward the positive electrode (anode) in the lower chamber, the leading ion is Cl− ( high mobility and high concentration); glycinate is the trailing ion (low mobility and low concentration). SDS-protein particles do not migrate freely at the border between the Cl− of the gel buffer and the Gly− of the cathode buffer. Friedrich Kohlrausch found that Ohm's law also applies to dissolved electrolytes. Because of the voltage drop between the Cl− and Glycine-buffers, proteins are compressed (stacked) into micrometer thin layers.[10] The boundary moves through a pore gradient and the protein stack gradually disperses due to a frictional resistance increase of the gel matrix. Stacking and unstacking occurs continuously in the gradient gel, for every protein at a different position. For a complete protein unstacking the polyacrylamide-gel concentration must exceed 16% T. The two-gel system of "Laemmli" is a simple gradient gel. The pH discontinuity of the buffers is of no significance for the separation quality, and a "stacking-gel" with a different pH is not needed.
Visualization
The most popular protein stain is Coomassie Brilliant Blue. It is an anionic dye, which non-specifically binds to proteins. Proteins in the gel are fixed by acetic acid and simultaneously stained. The excess dye incorporated into the gel can be removed by destaining with the same solution without the dye. The proteins are detected as blue bands on a clear background.
When more sensitive method than staining by Coomassie is needed silver staining is usually used. Silver staining is a sensitive procedure to detect trace amounts of proteins in gels, but can also visualize nucleic acid or polysaccharides.
Visualization methods without using a dye such as Coomassie and silver are available on the market. For example Bio-Rad Laboratories markets ”stain-free” gels for SDS-PAGE gel electrophoresis. Alternatively, reversible fluorescent dyes from Azure Biosystems such as AzureRed or Azure TotalStain Q can be used.
Similarly as in nucleic acid gel electrophoresis, tracking dye is often used. Anionic dyes of a known electrophoretic mobility are usually included in the sample buffer. A very common tracking dye is Bromophenol blue. This dye is coloured at alkali and neutral pH and is a small negatively charged molecule that moves towards the anode. Being a highly mobile molecule it moves ahead of most proteins.
Medical applications
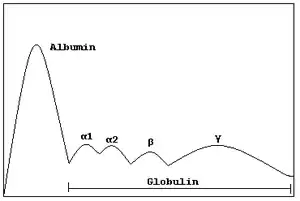

In medicine, protein electrophoresis is a method of analysing the proteins mainly in blood serum. Before the widespread use of gel electrophoresis, protein electrophoresis was performed as free-flow electrophoresis (on paper) or as immunoelectrophoresis.
Traditionally, two classes of blood proteins are considered: serum albumin and globulin. They are generally equal in proportion, but albumin as a molecule is much smaller and lightly, negatively-charged, leading to an accumulation of albumin on the electrophoretic gel. A small band before albumin represents transthyretin (also named prealbumin). Some forms of medication or body chemicals can cause their own band, but it usually is small. Abnormal bands (spikes) are seen in monoclonal gammopathy of undetermined significance and multiple myeloma, and are useful in the diagnosis of these conditions.
The globulins are classified by their banding pattern (with their main representatives):
- The alpha (α) band consists of two parts, 1 and 2:
- α1 - α1-antitrypsin, α1-acid glycoprotein.
- α2 - haptoglobin, α2-macroglobulin, α2-antiplasmin, ceruloplasmin.
- The beta (β) band - transferrin, LDL, complement
- The gamma (γ) band - immunoglobulin (IgA, IgD, IgE, IgG and IgM). Paraproteins (in multiple myeloma) usually appear in this band.
Normal present medical procedure involves determination of numerous proteins in plasma including hormones and enzymes, some of them also determined by electrophoresis. However, gel electrophoresis is mainly a research tool, also when the subject is blood proteins.
See also
References
- Schägger, H.; Jagow, G. (1991). "Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form". Anal. Biochem. 199 (2): 223–231. doi:10.1016/0003-2697(91)90094-A. PMID 1812789.
- Wittig, I.; Braun, H.P.; Schägger, H. (2006). "Blue native PAGE". Nat. Protoc. 1 (1): 418–428. doi:10.1038/nprot.2006.62. PMID 17406264.
- Wittig, I.; Schägger, H. (Nov 2005). "Advantages and limitations of clear-native PAGE". Proteomics. 5 (17): 4338–46. doi:10.1002/pmic.200500081. PMID 16220535. Archived from the original on 2013-01-05.
- Gavin P.D.; Devenish R.J.; Prescott M. (2003). "FRET reveals changes in the F1–stator stalk interaction during activity of F1F0-ATP synthase". Biochim Biophys Acta. 1607 (2–3): 167–79. doi:10.1016/j.bbabio.2003.09.013. PMID 14670607.
- Kastenholz, B. (2004). "Preparative native continuous polyacrylamide gel electrophoresis (PNC‐PAGE): an efficient method for isolating cadmium cofactors in biological systems". Protein Pept Lett. 37 (4): 657–65. doi:10.1081/AL-120029742. S2CID 97636537.
- Ornstein L (December 1964). "Disc Electrophoresis. I. Background and Theory". Annals of the New York Academy of Sciences. 121 (2): 321–349. Bibcode:1964NYASA.121..321O. CiteSeerX 10.1.1.140.7598. doi:10.1111/j.1749-6632.1964.tb14207.x. PMID 14240533.
- Davis BJ (December 1964). "Disc Electrophoresis. 2, Method and application to human serum proteins". Ann. N. Y. Acad. Sci. 121 (2): 404–427. Bibcode:1964NYASA.121..404D. doi:10.1111/j.1749-6632.1964.tb14213.x. PMID 14240539.
- Schägger H, von Jagow G (1987). "Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa". Anal. Biochem. 166 (2): 368–379. doi:10.1016/0003-2697(87)90587-2. PMID 2449095.
- Wiltfang J, Arold N, Neuhoff V (1991). "A new multiphasic buffer system for sodium dodecyl sulfate-polyacrylamide gel electrophoresis of proteins and peptides with molecular masses 100,000-1000, and their detection with picomolar sensitivity". Electrophoresis. 12 (5): 352–366. doi:10.1002/elps.1150120507. PMID 1718736.
- Kohlrausch F (1897). "Ueber Concentrations-Verschiebungen durch Electrolyse im Inneren von Lösungen und Lösungsgemischen". Annalen der Physik und Chemie. 62 (10): 209–239. Bibcode:1897AnP...298..209K. doi:10.1002/andp.18972981002.
- Minde DP (2012). "Determining biophysical protein stability in lysates by a fast proteolysis assay, FASTpp". PLOS ONE. 7 (10): e46147. Bibcode:2012PLoSO...746147M. doi:10.1371/journal.pone.0046147. PMC 3463568. PMID 23056252.