Peptide mass fingerprinting
Peptide mass fingerprinting (PMF) (also known as protein fingerprinting) is an analytical technique for protein identification in which the unknown protein of interest is first cleaved into smaller peptides, whose absolute masses can be accurately measured with a mass spectrometer such as MALDI-TOF or ESI-TOF.[1] The method was developed in 1993 by several groups independently.[2][3][4][5][6] The peptide masses are compared to either a database containing known protein sequences or even the genome. This is achieved by using computer programs that translate the known genome of the organism into proteins, then theoretically cut the proteins into peptides, and calculate the absolute masses of the peptides from each protein. They then compare the masses of the peptides of the unknown protein to the theoretical peptide masses of each protein encoded in the genome. The results are statistically analyzed to find the best match.
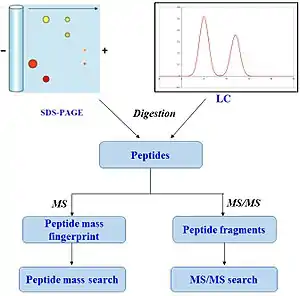
The advantage of this method is that only the masses of the peptides have to be known. Time-consuming de novo peptide sequencing is then unnecessary. A disadvantage is that the protein sequence has to be present in the database of interest. Additionally most PMF algorithms assume that the peptides come from a single protein.[7] The presence of a mixture can significantly complicate the analysis and potentially compromise the results. Typical for the PMF based protein identification is the requirement for an isolated protein. Mixtures exceeding a number of 2-3 proteins typically require the additional use of MS/MS based protein identification to achieve sufficient specificity of identification (6). Therefore, the typical PMF samples are isolated proteins from two-dimensional gel electrophoresis (2D gels) or isolated SDS-PAGE bands. Additional analyses by MS/MS can either be direct, e.g., MALDI-TOF/TOF analysis or downstream nanoLC-ESI-MS/MS analysis of gel spot eluates.[7][8]
Origins
Due to the long, tedious process of analyzing proteins, peptide mass fingerprinting was developed. Edman degradation was used in protein analysis, and it required almost an hour to analyze one amino acid residue.[9] SDS-PAGE was also used to separate proteins in very complex mixtures, which also employed methods of electroblotting and staining.[10] Then, bands would be extracted from the gel and sequenced, automatically. A recurring problem in the process was that interfering proteins would also purify with the protein of interest. The sequences of these interfering proteins were compiled into what came to known as the Dayhoff database.[11] Ultimately, having the sequences of these known protein contaminants in databases decreased instrument time and expenses involved in protein analysis.
Sample preparation
Protein samples can be derived from SDS-PAGE[7] or reversed phase HPLC, and are then subject to some chemical modifications. Disulfide bridges in proteins are reduced and cysteine amino acids are carbamidomethylated chemically or acrylamidated during the gel electrophoresis.
Then the proteins are cut into several fragments using proteolytic enzymes such as trypsin, chymotrypsin or Glu-C. A typical sample:protease ratio is 50:1. The proteolysis is typically carried out overnight and the resulting peptides are extracted with acetonitrile and dried under vacuum. The peptides are then dissolved in a small amount of distilled water or further concentrated and purified and are ready for mass spectrometric analysis.
Mass spectrometric analysis
The digested protein can be analyzed with different types of mass spectrometers such as ESI-TOF or MALDI-TOF. MALDI-TOF is often the preferred instrument because it allows a high sample throughput and several proteins can be analyzed in a single experiment, if complemented by MS/MS analysis. LC/ESI-MS and CE/ESI-MS are also great techniques for peptide mass fingerprinting.[12][13]
A small fraction of the peptide (usually 1 microliter or less) is pipetted onto a MALDI target and a chemical called a matrix is added to the peptide mix. Common matrices are Sinapinic acid, Alpha-Cyano-4-hydroxycinnamic acid, and 2,3-Dihydroxybenzoic acid. The matrix molecules are required for the desorption of the peptide molecules. Matrix and peptide molecules co-crystallize on the MALDI target and are ready to be analyzed. There is one predominantly MALDI-MS sample preparation technique, namely dried droplet technique.[14] The target is inserted into the vacuum chamber of the mass spectrometer and the desorption and ionisation of the polypeptide fragments is initiated by a pulsed laser beam which transfers high amounts of energy into the matrix molecules. The energy transfer is sufficient to promote the ionisation and transition of matrix molecules and peptides from the solid phase into the gas phase. The ions are accelerated in the electric field of the mass spectrometer and fly towards an ion detector where their arrival is detected as an electric signal. Their mass-to-charge ratio is proportional to their time of flight (TOF) in the drift tube and can be calculated accordingly.
Coupling ESI with capillary LC can separate peptides from protein digests, while obtaining their molecular masses at the same time.[15] Capillary electrophoresis coupled with ESI-MS is another technique; however, it works best when analyzing small amounts of proteins.[13]
Computational analysis
The mass spectrometric analysis produces a list of molecular weights of the fragments which is often called a peak list. The peptide masses are compared to protein databases such as Swissprot, which contain protein sequence information. Software performs in silico digests on proteins in the database with the same enzyme (e.g. trypsin) used in the chemical cleavage reaction. The mass of these peptide fragments is then calculated and compared to the peak list of measured peptide masses. The results are statistically analyzed and possible matches are returned in a results table.
References
- Clauser KR, Baker P, Burlingame AL (1999). "Role of accurate mass measurement (+/- 10 ppm) in protein identification strategies employing MS or MS/MS and database searching". Anal. Chem. 71 (14): 2871–82. doi:10.1021/ac9810516. PMID 10424174.
- Pappin DJ, Hojrup P, Bleasby AJ (1993). "Rapid identification of proteins by peptide-mass fingerprinting". Curr. Biol. 3 (6): 327–32. doi:10.1016/0960-9822(93)90195-T. PMID 15335725.
- Henzel WJ, Billeci TM, Stults JT, Wong SC, Grimley C, Watanabe C (1993). "Identifying proteins from two-dimensional gels by molecular mass searching of peptide fragments in protein sequence databases". Proc. Natl. Acad. Sci. U.S.A. 90 (11): 5011–5. Bibcode:1993PNAS...90.5011H. doi:10.1073/pnas.90.11.5011. PMC 46643. PMID 8506346.
- Mann M, Højrup P, Roepstorff P (1993). "Use of mass spectrometric molecular weight information to identify proteins in sequence databases". Biological Mass Spectrometry. 22 (6): 338–45. doi:10.1002/bms.1200220605. PMID 8329463.
- James P, Quadroni M, Carafoli E, Gonnet G (1993). "Protein identification by mass profile fingerprinting". Biochem. Biophys. Res. Commun. 195 (1): 58–64. doi:10.1006/bbrc.1993.2009. PMID 8363627.
- Yates JR, Speicher S, Griffin PR, Hunkapiller T (1993). "Peptide mass maps: a highly informative approach to protein identification". Anal. Biochem. 214 (2): 397–408. doi:10.1006/abio.1993.1514. PMID 8109726.
- Shevchenko A, Jensen ON, Podtelejnikov AV, Sagliocco F, Wilm M, Vorm O, Mortensen P, Shevchenko A, Boucherie H, Mann M (1996). "Linking genome and proteome by mass spectrometry: large-scale identification of yeast proteins from two dimensional gels". Proc. Natl. Acad. Sci. U.S.A. 93 (25): 14440–5. Bibcode:1996PNAS...9314440S. doi:10.1073/pnas.93.25.14440. PMC 26151. PMID 8962070.
- Wang W, Sun J, Nimtz M, Deckwer WD, Zeng AP (2003). "Protein identification from two-dimensional gel electrophoresis analysis of Klebsiella pneumoniae by combined use of mass spectrometry data and raw genome sequences". Proteome Science. 1 (1): 6. doi:10.1186/1477-5956-1-6. PMC 317362. PMID 14653859.
- Henzel, William J.; Watanabe, Colin; Stults, John T. (2003-09-01). "Protein identification: The origins of peptide mass fingerprinting". Journal of the American Society for Mass Spectrometry. 14 (9): 931–942. doi:10.1016/S1044-0305(03)00214-9. ISSN 1044-0305. PMID 12954162.
- Matsudaira, P. (1987-07-25). "Sequence from picomole quantities of proteins electroblotted onto polyvinylidene difluoride membranes". The Journal of Biological Chemistry. 262 (21): 10035–10038. ISSN 0021-9258. PMID 3611052.
- B C Orcutt; D G George; Dayhoff, and M. O. (1983). "Protein and Nucleic Acid Sequence Database Systems". Annual Review of Biophysics and Bioengineering. 12 (1): 419–441. doi:10.1146/annurev.bb.12.060183.002223. PMID 6347043.
- Moore, R. E.; Licklider, L.; Schumann, D.; Lee, T. D. (1998-12-01). "A microscale electrospray interface incorporating a monolithic, poly(styrene-divinylbenzene) support for on-line liquid chromatography/tandem mass spectrometry analysis of peptides and proteins". Analytical Chemistry. 70 (23): 4879–4884. doi:10.1021/ac980723p. ISSN 0003-2700. PMID 9852776.
- Whitmore, Colin D.; Gennaro, Lynn A. (2012-06-01). "Capillary electrophoresis-mass spectrometry methods for tryptic peptide mapping of therapeutic antibodies". Electrophoresis. 33 (11): 1550–1556. doi:10.1002/elps.201200066. ISSN 1522-2683. PMID 22736356.
- Thiede, Bernd (2005). "Peptide mass fingerprinting". Methods. 35 (3): 237–247. doi:10.1016/j.ymeth.2004.08.015. PMID 15722220.
- Dass, Chhabil (2007). Fundamentals of Contemporary Mass Spectrometry | Wiley Online Books. doi:10.1002/0470118490. ISBN 9780470118498.
External links
| Library resources about Peptide mass fingerprinting |