Host switch
In parasitology and epidemiology, a host switch (or host shift) is an evolutionary change of the host specificity of a parasite or pathogen. For example, the human immunodeficiency virus used to infect and circulate in non-human primates in West-central Africa, but switched to humans in the early 20th century.[1][2]
All symbiotic species, such as parasites, pathogens and mutualists, exhibit a certain degree of host specificity. This means that pathogens are highly adapted to infect a specific host - in terms of but not limited to receptor binding, countermeasures for host restriction factors and transmission methods. They occur in the body (or on the body surface) of a single host species or – more often – on a limited set of host species. In the latter case, the suitable host species tend to be taxonomically related, sharing similar morphology and physiology.[3]
Speciation is the creation of a new and distinct species through evolution and so unique differences exist between all life on earth. It goes without saying that dogs and birds are very different classes of animals – for one, dogs have fur coats and birds have feathers and wings. We therefore know that their fundamental biological makeup is as different as their physical appearance, this ranges from their internal cellular mechanisms to their response to infection, and so species-specific pathogens must overcome multiple host range barriers in order for their new host to support their infection.

Types of host switching
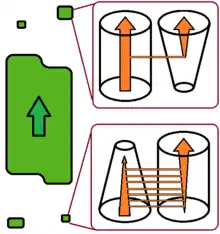
Recent studies have proposed to discriminate between two different types of evolutionary change in host specificity.[5][6]
According to this view, host switch can be a sudden and accidental colonization of a new host species by a few parasite individuals capable of establishing a new and viable population there. After a switch of this type, the new population is more-or-less isolated from the population on the donor host species. The new population does not affect the further fate of the conspecific parasites on the donor host, and may finally lead to parasite speciation. This type of switch is more likely to target an increasing host population that harbours a relatively poor parasite/pathogen fauna, such as the pioneer populations of invasive species. The switch of HIV to the human host is of this type.
Alternatively, in the case of a multi-host parasite host-shift may occur as a gradual change of the relative role of one host species, which becomes primary rather than secondary host. The former primary host slowly becomes a secondary host, or may even, eventually, be totally abandoned. This process is slower and more predictable, and does not increase parasite diversity. It will typically occur in a shrinking host population harbouring a parasite/pathogen fauna which is relatively rich for the host population size.
Host switching features
Reason for host switch events
All diseases have an origin. Some disease circulate in human populations and are already known to epidemiologists, but evolution of the disease can result in a new strain of this disease emerging that makes it stronger - for example, multi-drug resistant tuberculosis. In other cases, diseases can be discovered that have not previously been observed or studied. These can emerge due to host switch events allowing the pathogen to evolve to become human-adapted and are only discovered due to an infection outbreak.
A pathogen that switches host emerges as a new form of the virus capable of circulating within a new population. Diseases that emerge in this sense can occur more often through human over exposure to the wildlife. This can be as a result of urbanisation, deforestation, destruction of wildlife habitats and changes to agricultural practise. The more exposure humans have to the wild, the more spillover infections occur and pathogens are exposed to human-specific selection pressures. The pathogen is therefore driven towards specific-specific adaptation and is more likely to gain the necessary mutations to jump the species barrier and become human-infective.
Host switch and pathogenicity
The problem with diseases emerging in new species is that the host population will be immunologically naïve. This means that the host has never been previously exposed to the pathogen and has no pre-existing antibodies or protection from the infection. This make host switching dangerous and can result in more pathogenic infections. The pathogen is not adapted to surviving in this new host and this imbalance of coevolutionary history may result in aggressive infections. However, this balance must be brought under control for the pathogen to maintain its infection in the new host and not burn through the population.
Stages of host switch
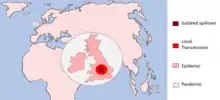
A pathogen undergoing a host switch is driven by selection pressures to acquire the necessary changes allowing for survival and transmission in the new host species. According to a 2008 Microbiology and Molecular Biology Review,[7] this process of host switching can be defined by three stages:
- Isolated infection
- -An isolated infection of a new host with no further infection within the new species
- -Spillovers into dead-end hosts
- Local spillovers
- -Spillover events that cause small chains of local transmission
- -Sustained epidemic transmission of the pathogen within the new host species
- -Global spread of the disease infection
Exposure to new environments and host species is what allows pathogens to evolve. The early isolated infection events exposes the pathogen to the selection pressure of survival in that new species of which some will eventually adapt to. This gives raise to pathogens with the primary adaptations allowing the smaller outbreaks within this potential new host, increasing exposure and driving further evolution. This gives rise to complete host adaptation and the capability for a larger epidemic and the pathogen can sustainably survive in its new host – i.e. host switch. Sufficiently adapted pathogens may also reach pandemic status meaning the disease has infected the whole country or spread around the world.
Zoonosis and spillover
A zoonosis is a specific kind of cross-species infection in which diseases are transmitted from vertebrate animals to humans. An important feature of a zoonotic disease is they originate from animal reservoirs which are essential to the survival of zoonotic pathogens.[8] They naturally exist in animal populations asymptomatically - or causing mild disease - making it challenging to find the natural host (disease reservoir) and impossible to eradicate as the virus will always continue to live in wild animal species.
Those zoonotic pathogens that permanently make the jump from vertebrate animals to human populations have performed a host switch and thus can continue to survive as they are adapted to transmission in human populations. However, not all zoonotic infections complete the host switch and only exist as smaller isolated events. These are known as spillovers. This means that humans can become infected from an animal pathogen, but it does not necessarily take hold and become a human transmitted disease that circulates in human populations. This is because the host switch adaptations required to make the pathogen sustainable and transmissible in the new host does not occur.
Some cross-species transmission events are important as they can show that a pathogen is getting closer to epidemic/pandemic potential. Small epidemics show that the pathogen is getting more adapted to human transmission and gaining stability to exist in the human population. However, there are some pathogens that do not possess this ability to spread between humans. This is the case for spillover events such as rabies. Humans infected from the bite of rabid animals do not tend to pass on the disease and so are classed as dead-end hosts.[9]
An extensive list of zoonotic infections can be found at Zoonosis.
Case studies
The following pathogens are examples of diseases that have crossed the species barrier into the human population and highlight the complexity of the switch.
Influenza
| Influenza | |
|---|---|
 | |
| Influenza virus has an 8 segment genome encapsulated within the viral matrix surrounded by the viral capsule containing the hemaglutinnin and neuraminidase proteins. | |
| Virus classification | |
| Group: | Group V ((−)ssRNA) |
| Family: | |
| Genus: | Alphainfluenzavirus |
| Species: | Influenza a virus |
Influenza - also known as the flu - is one of the most well-known viruses that continues to pose a huge burden on today's health care systems and is the most common cause of human respiratory infections.[10] Influenza is an example of how a virus can continuously jump the species barrier in multiple isolated instances over time creating different human infecting strains circulating our populations - for example, H1N1, H5N1 and H7N9. These host switch events create pandemic strains that eventually transition into seasonal flu that annually circulates in the human population in colder months.
Influenza A viruses (IAVs) are classified by two defining proteins. These proteins are present in all influenza viral strains but small differences allow for differentiation of new strains. These identifiers are:
- hemagglutinin (HA)
- neuraminidase (NA)
IAVs naturally exist in wild birds without causing disease or symptoms. These birds, especially waterfowl and shore birds, are the reservoir host of the majority of IAVs with these HA and NA protein antigens.[11] From these animals, the virus spills into other species (e.g. pigs, humans, dogs [10]) creating smaller scale infections until the virus has acquired significant mutations to spread and maintain itself in another species. The RNA polymerase enzyme of influenza has low level accuracy due to a lack of a proofreading mechanism and therefore has a high error rate in terms of genetic replication.[10] Because of this, influenza has the capacity to mutate frequently dependent of the current selection pressures and has the capability to adapt to surviving in different host species.
Transmission and infection methods
Comparing IAVs in birds and humans, one of the main barriers to host switching is the type of cells the virus can recognise and bind to (cell tropism) in order to initiate infection and viral replication. An avian influenza virus is adapted to binding to the gastrointestinal tract of birds.[11] In bird populations, the virus is shed from the excretory system into the water and ingested by other birds to colonise their guts. This is not the case in humans as influenza, in this species, produces a respiratory infection. The virus here binds to respiratory tissue and is transmitted through breathing, talking and coughing, therefore the virus has to adapt in order to switch to the human host from avian populations. Additionally, the respiratory tract is mildly acidic and so the virus must also mutate to overcome these conditions in order to successfully colonise mammalian lungs and respiratory tracts. Acidic conditions are a trigger for viral uncoating as it is normally a sign the virus has penetrated a cell, however premature uncoating will result in virus exposure to the immune system leading killing of the virus.[12]
Host receptor binding
IAVs binds to host cells using the HA protein. These proteins recognise sialic acid that reside on the terminal regions of external glycoproteins on host cell membranes. However, HA proteins have different specificities for isomers of sialic acid depending on which species the IAV is adapted for. IAVs adapted for birds recognise α2-3 sialic acid isomers whereas human adapted IAV HAs bind to α2-6 isomers.[10] These are the isomers of sialic acid mostly present in the regions of the host that each IAVs infected respectively - i.e. the gastrointestinal tract of birds and the respiratory tract of humans. Therefore, in order to commit to a host switch, the HA specificity must mutate to the substrate receptors of the new host.
In the final stages of infection, the HA proteins are cleaved to activate the virus.[10] Certain hemagglutinin subtypes (H5 and H7) have the capacity to obtain additional mutations. These exist at the HA activation cleavage site which changes the HA specificity. This results in a broadening of the range of protease enzymes that can bind to and activate the virus. Therefore, this makes the virus more pathogenic and can make IAV infections more aggressive.[10]
Polymerase action
Successfully binding to different host tissue is not the only requirement of host switch for influenza A. The influenza genome is replicated using the virus RNA-dependent RNA polymerase but it must adapt to use host specific cofactors in order to function.[13] The polyermase is a heterotrimeric complex and consists of 3 major domains: PB1, PB2 and PA. Each plays their own role in replication of the viral genome but PB2 is an important factor in the host range barrier as it interacts with host cap proteins.[10] Specifically, residue 627 of the PB2 unit shows to play a defining role in the host switch from avian to human adapted influenza strains. In IAVs, the residue at position 627 is glutamic acid (E) whereas in mammal infecting influenza, this residue is mutated to lysine (K).[13][14] Therefore, the virus must undergo a E627K mutation in order to perform a mammalian host switch. This region surrounding residue 627 forms a cluster protruding from the enzyme core. With lysine, this PB2 surface region can form a basic patch enabling host cofactor interaction, whereas the glutamic acid residue found in IAVs disrupt this basic region and subsequent interactions.[13]
Host cofactor
The cellular protein ANP32A has been shown to account for contrasting levels of avian influenza interaction efficiency with different host species.[13][15] The key difference between ANP32A is that the avian form contains an additional 33 amino acids than the mammalian form.[15] When mammals cells are infected with avian IAVs, the polymerase enzyme efficiency is sub-optimal as the avian virus is not adapted to surviving in mammalian cells. However, when that mammalian cell contains the avian ANP32A protein, viral replication is mostly restored,[15] showing that the ANP32A is likely to positively interact and optimise the polymerase action. Mutations in the PB2 making the influenza mammal-adapted allow for the interaction between the viral polymerase and the mammalian ANP32A protein and therefore essential for the host switch.
Summary
There are many factor that determine a successful influenza host switch from avian to a mammalian host:
- Stability in the mildly acidic mammalian respiratory tract
- Recognition of mammalian sialic acid by HA receptors
- PB2 E627K mutation in the viral polymerase to allow for interaction with mammalian ANP32A for optimal viral replication
Each factor has a role to play and so the virus must acquire them all in order to undergo the host switch. This is a complex process and requires time for the virus to sufficiently adapt and mutate. Once each mutation has been achieved, the virus can infect human populations and has the potential to reach pandemic levels. However, this is dependent on virulence and rate of transmission and host switching will change these parameters of viral infection.
HIV
| HIV | |
|---|---|
 | |
| The human immunodeficiency viruses (both HIV1 and HIV2) exhibited evolutionary switches from non-human primates to humans. | |
| Virus classification | |
| Group: | Group VII (dsDNA-RT) |
| Family: | |
| Subfamily: | |
| Genus: | |
| Species: | |
HIV is the human immunodeficiency virus and attacks cells of the immune system depleting the body's defence against incoming pathogens. In particular, HIV infects CD4+ T helper lymphocytes, a cell involved in the organisation and coordination of the immune response. This means that the body can recognise incoming pathogens but cannot trigger their defences against them.[16] When HIV sufficiently diminishes the immune system, it causes a condition known as acquired immunodeficiency syndrome or AIDS characterised by severe weight loss, fever, swollen lymph nodes and susceptibility to other severe infections [17]
HIV is a type of lentivirus of which two types are known to cause AIDS: HIV-1 and HIV-2,[16][18] both of which jumped into the human population from numerous cross-species transmission events by the equivalent disease in primates known as simian immunodeficiency virus (SIV). SIVs are found in many different primate species, including chimpanzees and mandrills found in sub-Saharan Africa, and for the most part are largely non pathogenic[18] HIV-1 and HIV-2 having similar features but are antigenically different and so are classed as different types of HIV.[18] Most transmission events are unsuccessful in switching its host, however in the context of HIV-1, four distinct forms emerged categorised as groups M, N, O and P of which group M is associated with pandemic HIV-1 and accounts for the majority of global cases. Each type is proposed to have emerged through bush-meat hunting and the exposure to the body fluids of infected primates,[18] including blood.
Gag-30
Host-specific selection pressures would bring about a change in the viral proteome of HIVs to suit the new host and therefore these regions would not be conserved when compared to SIVs. Through these viral proteomic comparisons, the viral matrix protein Gag-30 was identified as having differing amino acids at position 30. This amino acid is conserved as a methionine in SIVs but mutated to an arginine or lysine in HIV-1 groups M, N and O,[18][19] suggesting a strong selection pressure in the new host. This observation was supported by other data including the fact that this mutation was reversed when HIV-1 was used to infect primates meaning that the arginine or lysine converted back to the methionine originally observed in SIVs.[19] This reinforces the idea of the strong, opposing host-specific selection pressure between humans and primates. Additionally, it was observed that methionine containing viruses replicated more efficiently in primates and arginine/lysine containing viruses in humans.[19] This is evidence of the reason behind the mutation (optimal levels of replication in host CD4+ T lymphocytes), however the exact function and action of the position 30 amino acid is unknown.
Tetherin countermeasures

Tetherin is a defence protein in the innate immune response whose production is activation by interferon. Tetherin specifically inhibits the infective capabilities of HIV-1 by blocking its release from the cells it infects.[20] This prevents the virus from leaving to infect more cells and halts the progression of the infection giving the host defences time to destroy the viral-infected cells. Adapted viruses tend to have countermeasures to defend themselves against tetherin normally through degradation through specific regions of the protein. These anti-tetherin techniques are different between SIVs and HIV-1 showing that tetherin interaction is a host range restriction that must be overcome to enable a primate-human host switch. SIVs use the Nef protein to remove tetherin from the cell membrane whereas HIV-1 uses the Vpu protein degrade the defence protein.[18]
Tetherin is a conserved viral-defence mechanism across species but its exact sequence and structure shows some differences. The regions making up tetherin include the cytoplasmic region, transmembrane region, a coiled-coiled extracellular domain and a GPI anchor;[18] however, human tetherin defers to other primates by having a deletion in the cytoplasmic region.[21] This incomplete cytoplasmic domain renders Nef proteins found in SIVs ineffective as an anti-tetherin response in humans and so in order to switch from non-human primates to a human host, the SIV must activate the Vpu protein which instead blocks tetherin through interaction with the conserved transmembrane region.[21]
Summary
The two factors that are involved in the host range barrier for SIV to HIV viruses are:
- Gag-30 protein - specifically the amino acid at position 30
- The use of Nef or Vpu proteins as an anti-tetherin defence
Only a SIV virus containing both mutations of the Gag-30 protein and acquisition of the Vpu anti-tetherin protein will be able to undergo a host switch from primates to humans and become a HIV. This evolutionary adaptations allows the virus to acquire optimal levels of polymerase action in human infected cells and the ability to prevent destruction of the virus by tetherin.
References
- Sharp PM, Hahn BH (September 2011). "Origins of HIV and the AIDS pandemic". Cold Spring Harbor Perspectives in Medicine. 1 (1): a006841. doi:10.1101/cshperspect.a006841. PMC 3234451. PMID 22229120.
- Faria NR, Rambaut A, Suchard MA, Baele G, Bedford T, Ward MJ, et al. (October 2014). "HIV epidemiology. The early spread and epidemic ignition of HIV-1 in human populations". Science. 346 (6205): 56–61. Bibcode:2014Sci...346...56F. doi:10.1126/science.1256739. PMC 4254776. PMID 25278604.
- Poulin R (2006). Evolutionary Ecology of Parasites. Princeton University Press.
- Reed DL, Light JE, Allen JM, Kirchman JJ (March 2007). "Pair of lice lost or parasites regained: the evolutionary history of anthropoid primate lice". BMC Biology. 5: 7. doi:10.1186/1741-7007-5-7. PMC 1828715. PMID 17343749.
- Rozsa L, Tryjanowski P, Vas Z (2015). "Under the changing climate: how shifting geographic distributions and sexual selection shape parasite diversification" (PDF). In Morand S, Krasnov B, Littlewood T (eds.). Parasite diversity and diversification: evolutionary ecology meets phylogenetics. Cambridge University Press. pp. 58–76. ISBN 9781107037656.
- Forro B, Eszterbauer E (June 2016). "Correlation between host specificity and genetic diversity for the muscle-dwelling fish parasite Myxobolus pseudodispar: examples of myxozoan host-shift?" (PDF). Folia Parasitologica. 63: 019. doi:10.14411/fp.2016.019. PMID 27311917.
- Parrish CR, Holmes EC, Morens DM, Park EC, Burke DS, Calisher CH, et al. (September 2008). "Cross-species virus transmission and the emergence of new epidemic diseases". Microbiology and Molecular Biology Reviews. 72 (3): 457–70. doi:10.1128/MMBR.00004-08. PMC 2546865. PMID 18772285.
- "WHO | Zoonoses". WHO.
- Fooks AR, Cliquet F, Finke S, Freuling C, Hemachudha T, Mani RS, et al. (November 2017). "Rabies". Nature Reviews. Disease Primers. 3 (1): 17091. doi:10.1038/nrdp.2017.91. PMID 29188797.
- Taubenberger JK, Kash JC (June 2010). "Influenza virus evolution, host adaptation, and pandemic formation". Cell Host & Microbe. 7 (6): 440–51. doi:10.1016/j.chom.2010.05.009. PMC 2892379. PMID 20542248.
- Lewis DB (February 2006). "Avian flu to human influenza". Annual Review of Medicine. 57 (1): 139–54. doi:10.1146/annurev.med.57.121304.131333. PMID 16409141.
- Zaraket H, Bridges OA, Duan S, Baranovich T, Yoon SW, Reed ML, et al. (September 2013). "Increased acid stability of the hemagglutinin protein enhances H5N1 influenza virus growth in the upper respiratory tract but is insufficient for transmission in ferrets". Journal of Virology. 87 (17): 9911–22. doi:10.1128/JVI.01175-13. PMC 3754100. PMID 23824818.
- Nilsson BE, Te Velthuis AJ, Fodor E (April 2017). "Role of the PB2 627 Domain in Influenza A Virus Polymerase Function". Journal of Virology. 91 (7). doi:10.1128/JVI.02467-16. PMC 5355620. PMID 28122973.
- Subbarao EK, London W, Murphy BR (April 1993). "A single amino acid in the PB2 gene of influenza A virus is a determinant of host range". Journal of Virology. 67 (4): 1761–4. doi:10.1128/JVI.67.4.1761-1764.1993. PMC 240216. PMID 8445709.
- Long JS, Giotis ES, Moncorgé O, Frise R, Mistry B, James J, et al. (January 2016). "Species difference in ANP32A underlies influenza A virus polymerase host restriction". Nature. 529 (7584): 101–4. Bibcode:2016Natur.529..101L. doi:10.1038/nature16474. PMC 4710677. PMID 26738596.
- Whiteside A (2016). HIV & AIDS: A Very Short Introduction (2 ed.). 198 Madison Avenue, New York, NY 10016, United States of America: Oxford University Press. pp. 168 pages. ISBN 9780191040962.CS1 maint: location (link)
- "HIV/AIDS". www.who.int.
- Sharp PM, Hahn BH (September 2011). "Origins of HIV and the AIDS pandemic". Cold Spring Harbor Perspectives in Medicine. 1 (1): a006841. doi:10.1101/cshperspect.a006841. PMC 3234451. PMID 22229120.
- Wain LV, Bailes E, Bibollet-Ruche F, Decker JM, Keele BF, Van Heuverswyn F, et al. (August 2007). "Adaptation of HIV-1 to its human host". Molecular Biology and Evolution. 24 (8): 1853–60. doi:10.1093/molbev/msm110. PMC 4053193. PMID 17545188.
- Perez-Caballero D, Zang T, Ebrahimi A, McNatt MW, Gregory DA, Johnson MC, Bieniasz PD (October 2009). "Tetherin inhibits HIV-1 release by directly tethering virions to cells". Cell. 139 (3): 499–511. doi:10.1016/j.cell.2009.08.039. PMC 2844890. PMID 19879838.
- Zhang F, Wilson SJ, Landford WC, Virgen B, Gregory D, Johnson MC, et al. (July 2009). "Nef proteins from simian immunodeficiency viruses are tetherin antagonists". Cell Host & Microbe. 6 (1): 54–67. doi:10.1016/j.chom.2009.05.008. PMC 2852097. PMID 19501037.
