T helper cell
The T helper cells (Th cells), also known as CD4+ cells or CD4-positive cells, are a type of T cell that play an important role in the immune system, particularly in the adaptive immune system. As their name suggests, they "help" the activity of other immune cells by releasing cytokines, small protein mediators that alter the behavior of target cells that express receptors for those cytokines. These cells help to polarize the immune response into the appropriate kind depending on the nature of the immunological insult (virus vs. extracellular bacterium vs. intracellular bacterium vs. helminth vs. fungus vs. protist). They are generally considered essential in B cell antibody class switching, breaking cross-tolerance in dendritic cells, in the activation and growth of cytotoxic T cells, and in maximizing bactericidal activity of phagocytes such as macrophages and neutrophils.
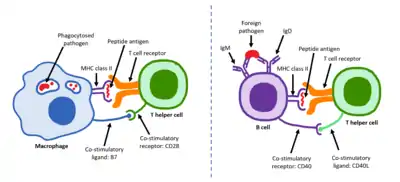
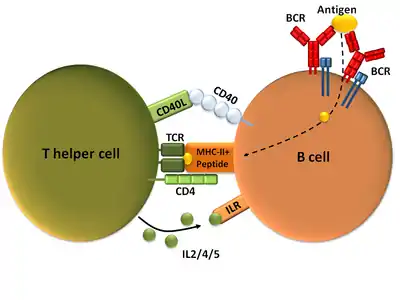
Mature Th cells express the surface protein CD4 and are referred to as CD4+ T cells. Such CD4+ T cells are generally treated as having a pre-defined role as helper T cells within the immune system. For example, when an antigen-presenting cell displays a peptide antigen on MHC class II proteins, a CD4+ cell will aid those cells through a combination of cell to cell interactions (e.g. CD40 (protein) and CD40L) and through cytokines.
CD154, also called CD40 ligand or CD40L, is a cell surface protein that mediates T cell helper function in a contact-dependent process[1] and is a member of the TNF superfamily of molecules. It binds to CD40 on antigen-presenting cells (APC), which leads to many effects depending on the target cell type. CD154 acts as a costimulatory molecule and is particularly important on a subset of T cells called T follicular helper cells (TFH cells).[2] On TFH cells, CD154 promotes B cell maturation and function by engaging CD40 on the B cell surface and therefore facilitating cell-cell communication.[3] A defect in this gene results in an inability to undergo immunoglobulin class switching and is associated with hyper IgM syndrome.[4] Absence of CD154 also stops the formation of germinal centers and therefore prohibits antibody affinity maturation, an important process in the adaptive immune system that allows for the generation of high-affinity antibodies that protect against future challenge by pathogens.
The importance of helper T cells collectively can be seen from HIV, a virus that primarily infects CD4+ T cells (but is capable of infecting other important cells of the immune system, like macrophages that express CD4). In the advanced stages of HIV infection, loss of functional CD4+ T cells leads to the symptomatic stage of infection known as the acquired immunodeficiency syndrome (AIDS). When HIV is detected early in blood or other bodily fluids, proper adherence to antiretroviral therapy will prevent the progression of HIV into AIDS and allow the body to naturally restore its own CD4 cell count (a small proportion of individuals is able to achieve prolonged control of viral load without significant decline in T cell levels over time without the aid of antiretroviral therapy, who are termed "elite controllers" or "long-term nonprogressors"). HIV is an example of a secondary immunodeficiency disorder. Primary immunodeficiencies are genetic conditions which result in immunological defects that prevent adequate clearance of infections and T cell deficiencies are particularly devastating. One example is SCID (severe combined immunodeficiency) which has many causes and varies in its precise phenotype depending on the cause. While most forms of SCID result in absence of both T and B cell lineages, atypical SCID is characterized (in part) by a normal level of B cells but profound T cell deficiency. However, because of the deficiency of helper T cells, the B cells have profoundly impaired function.[5]
It is critical to understand that viewing Th cells as a monolithic immunological entity is fallacious because they are extremely diverse in terms of function and their interaction with partner cells (this is elaborated upon below). In general, mature naive T cells (those which have passed through the checkpoints of development in the thymus but have not yet encountered their cognate antigen) are stimulated by professional antigen presenting cells to acquire an effector module. These are defined by the presence of a lineage-determining (or lineage-specifying) transcription factor (also called master regulator, though the term has been criticized for being too reductive).[6] The loss of function in a lineage specifying transcription factor results in the absence of the corresponding class of helper T cell which can be devastating for the health of the host. For example, conventional, regulatory T cells (Treg cells) are defined by the expression of FoxP3 (Forkhead Box P3) and the loss of function of this transcription factor results in a condition called IPEX (immunodysregulation polyendocrinopathy enteropathy X-linked) syndrome which is characterized by out-of-control immune responses that cause severe tissue damage as well as fulminant autoimmune disease (note that this is autoimmune disease and not merely the presence of autoreactivity) because this population of helper T cells is not present to dampen them. In some cases, a loss-of-function mutation can occur more upstream of the lineage-specifying transcription factor. For example, in Hyper IgE syndrome there is a mutation in the STAT3 gene which is responsible for inducing the RORγT transcription factor in response to the combination of TGF-β and either IL-6 or IL-21 which specifies TH17 cells. As a result of the absence of these cells patients have severe fungal infections and have difficulty responding to pyogenic bacterial pathogens (though they also have other immunological defects because STAT3 is involved in other signaling pathways as well).[7] STAT3 deficiency also compromises the ability to generate TFH cells.[8]
Activation of naive helper T cells
Following T cell development in the thymus, these cells (termed recent thymic emigrants (RTE)) egress from the thymus and home to secondary lymphoid organs (SLO; spleen and lymph nodes). Of note, only a very small minority of T cells egresses from the thymus (estimates commonly range from 1-5% but some experts feel even this is generous).[9] Maturation of RTE in SLO results in the generation of mature naive T cells (naïve meaning they have never been exposed to the antigen that they are programmed to respond to), but naive T cells now lack or have downregulated (reduced) expression of the RTE-related surface markers, such as CD31, PTK7, Complement Receptor 1 and 2 (CR1, CR2) and the production of interleukin 8 (IL-8).[10][11] Like all T cells, they express the T cell receptor-CD3 complex. The T cell receptor (TCR) consists of both constant and variable regions. The variable region determines what antigen the T cell can respond to. CD4+ T cells have TCRs with an affinity for Class II MHC, and CD4 is involved in determining MHC affinity during maturation in the thymus. Class II MHC proteins are generally only found on the surface of professional antigen-presenting cells (APCs). Professional antigen-presenting cells are primarily dendritic cells, macrophages and B cells, although dendritic cells are the only cell group that expresses MHC Class II constitutively (at all times). Some APCs also bind native (or unprocessed) antigens to their surface, such as follicular dendritic cells (these are not the same type of cells as the dendritic cells of the immune system but rather have a non-hematopoietic origin, and in general lack MHC Class II, meaning they are not true professional antigen-presenting cells; however, follicular dendritic cells may acquire MHC Class II proteins via exosomes that become attached to them[12]). T cells require antigens to be processed into short fragments which form linear epitopes on MHC Class II (in the case of helper T cells because they express CD4) or MHC class I (in the case of cytotoxic T cells which express CD8). MHC Class II binding pockets are flexible with respect to the length of the peptides they hold. Generally, there are 9 core amino acid residues with several flanking amino acids which form a length of about 12-16 amino acids total[13] but have been known to hold as many as 25 amino acids.[14] By comparison, MHC Class I proteins are usually 9-10 peptides long.[15] The activation of naive T cells is commonly explained in terms of the 3-signal model, elaborated upon below.[16]
Activation (signal 1)
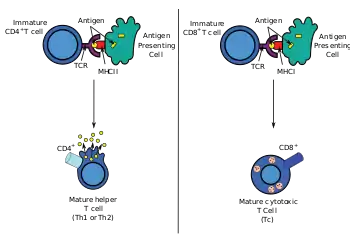
During an immune response, professional antigen-presenting cells (APCs) endocytose antigens (typically bacteria or viruses), which undergo processing, then travel from the infection site to the lymph nodes. Typically, the APC responsible is a dendritic cell. If the antigen expresses appropriate molecular patterns (sometimes known as signal 0), it can induce maturation of the dendritic cell which results in enhanced expression of costimulatory molecules needed to activate T cells (see signal 2)[17] and MHC Class II.[18] Once at the lymph nodes, the APCs begin to present antigen peptides that are bound to Class II MHC, allowing CD4+ T cells that express the specific TCRs against the peptide/MHC complex to activate.
When a Th cell encounters and recognizes the antigen on an APC, the TCR-CD3 complex binds strongly to the peptide-MHC complex present on the surface of professional APCs. CD4, a co-receptor of the TCR complex, also binds to a different section of the MHC molecule. It is estimated that approximately 50 of these interactions are required for the activation of a helper T cell and assemblies known as microclusters have been observed forming between the TCR-CD3-CD4 complexes of the T cell and the MHC Class II proteins of the dendritic cell at the zone of contact. When these all come together, the CD4 is able to recruit a kinase called Lck which phosphorylates immunotyrosine activation motifs (ITAMs) present on the CD3 gamma, delta, epsilon, and zeta chains. The protein ZAP-70 can bind these phosphorylated ITAMs via its SH2 domain and then itself becomes phosphorylated, wherein it orchestrates the downstream signaling required for T cell activation. Lck activation is controlled by the opposing actions of CD45 and Csk.[19] CD45 activates Lck by dephosphorylating a tyrosine in its C-terminal tail, while Csk phosphorylates Lck at that site. The loss of CD45 produces a form of SCID because failure to activate Lck prevents appropriate T cell signaling. Memory T cells also make use of this pathway and have higher levels of Lck expressed and the function of Csk is inhibited in these cells.[20]
The binding of the antigen-MHC to the TCR complex and CD4 may also help the APC and the Th cell adhere during Th cell activation, but the integrin protein LFA-1 on the T cell and ICAM on the APC are the primary molecules of adhesion in this cell interaction.
It is unknown what role the relatively bulky extracellular region of CD45 plays during cell interactions, but CD45 has various isoforms that change in size depending on the Th cell's activation and maturation status. For example, CD45 shortens in length following Th activation (CD45RA+ to CD45RO+), but whether this change in length influences activation is unknown. It has been proposed that the larger CD45RA may decrease the accessibility of the T cell receptor for the antigen-MHC molecule, thereby necessitating an increase in the affinity (and specificity) of the T cell for activation. However, once the activation has occurred, CD45 shortens, allowing easier interactions and activation as an effector T helper cell.
Survival (signal 2)
Having received the first TCR/CD3 signal, the naïve T cell must activate a second independent biochemical pathway, known as Signal 2. This verification step is a protective measure to ensure that a T cell is responding to a foreign antigen. If this second signal is not present during initial antigen exposure, the T cell presumes that it is auto-reactive. This results in the cell becoming anergic (anergy is generated from the unprotected biochemical changes of Signal 1). Anergic cells will not respond to any antigen in the future, even if both signals are present later on. These cells are generally believed to circulate throughout the body with no value until they undergo apoptosis.
The second signal involves an interaction between CD28 on the CD4+ T cell and the proteins CD80 (B7.1) or CD86 (B7.2) on the professional APCs. Both CD80 and CD86 activate the CD28 receptor. These proteins are also known as co-stimulatory molecules.
Although the verification stage is necessary for the activation of naïve helper T cells, the importance of this stage is best demonstrated during the similar activation mechanism of CD8+ cytotoxic T cells. As naïve CD8+ T cells have no true bias towards foreign sources, these T cells must rely on the activation of CD28 for confirmation that they recognise a foreign antigen (as CD80/CD86 is only expressed by active APC's). CD28 plays an important role in decreasing the risk of T cell auto-immunity against host antigens.
Once the naïve T cell has both pathways activated, the biochemical changes induced by Signal 1 are altered, allowing the cell to activate instead of undergoing anergy. The second signal is then obsolete; only the first signal is necessary for future activation. This is also true for memory T cells, which is one example of learned immunity. Faster responses occur upon reinfection because memory T cells have already undergone confirmation and can produce effector cells much sooner.
Differentiation (signal 3)
Once the two-signal activation is complete the T helper cell (Th) then allows itself to proliferate. It achieves this by releasing a potent T cell growth factor called interleukin 2 (IL-2) which acts upon itself in an autocrine fashion. Activated T cells also produce the alpha sub-unit of the IL-2 receptor (CD25 or IL-2R), enabling a fully functional receptor that can bind with IL-2, which in turn activates the T cell's proliferation pathways.
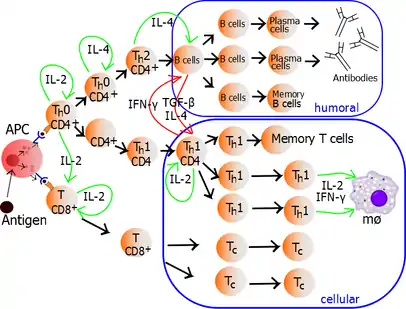
The autocrine or paracrine secretion of IL-2 can bind to that same Th cell or neighboring Th's via the IL-2R thus driving proliferation and clonal expansion. The Th cells receiving both signals of activation and proliferation will then become Th0 cells (T helper 0) cell that secrete IL-2, IL-4 and interferon gamma (IFN-γ). The Th0 cells will then differentiate into Th1 or Th2 cells depending on cytokine environment. IFN-γ drives Th1 cell production while IL-10 and IL-4 inhibit Th1 cell production. Conversely, IL-4 drives Th2 cell production and IFN-γ inhibits Th2 cells. These cytokines are pleiotropic and carry out many other functions of the immune response.
Effector function
In 1991, three groups reported discovering CD154, which is the molecular basis of T cell helper function. Seth Lederman at Columbia University generated a murine monoclonal antibody, 5c8 that inhibited contact-dependent T cell helper function in human cells which characterized the 32 kDa surface protein transiently expressed on CD4+ T cells.[1] Richard Armitage at Immunex cloned a cDNA encoding CD154 by screening an expression library with CD40-Ig.[21] Randolph Noelle at Dartmouth Medical School generated an antibody that bound a 39 kDa protein on murine T cells and inhibited helper function.[22]
Determination of the effector T cell response
Helper T cells are capable of influencing a variety of immune cells, and the T cell response generated (including the extracellular signals such as cytokines) can be essential for a successful outcome from infection. In order to be effective, helper T cells must determine which cytokines will allow the immune system to be most useful or beneficial for the host. Understanding exactly how helper T cells respond to immune challenges is currently of major interest in immunology, because such knowledge may be very useful in the treatment of disease and in increasing the effectiveness of vaccination.
Th1/Th2 model
Proliferating helper T cells that develop into effector T cells differentiate into two major subtypes of cells known as Th1 and Th2 cells (also known as Type 1 and Type 2 helper T cells, respectively).
Th1 helper cells lead to an increased cell-mediated response, typically against intracellular bacteria and protozoa. They are triggered by the polarising cytokine IL-12 and their effector cytokines are IFN-γ and IL-2. The main effector cells of Th1 immunity are macrophages as well as CD8 T cells, IgG B cells, and IFN-γ CD4 T cells. The key Th1 transcription factors are STAT4 and T-bet. IFN-γ secreted by CD4 T cells can activate macrophages to phagocytose and digest intracellular bacteria and protozoa. In addition, IFN-γ can activate iNOS (inducible nitric oxide synthase) to produce nitric oxide free radicals to directly kill intracellular bacteria and protozoa. Th1 overactivation against autoantigens will cause Type IV or delayed-type hypersensitivity reaction. Tuberculin reaction or Type 1 diabetes belong to this category of autoimmunity.[23]
Th2 helper cells lead to a humoral immune response, typically against extracellular parasites including helminths. They are triggered by the polarising cytokines IL-4 and IL-2, and their effector cytokines are IL-4, IL-5, IL-9, IL-10, IL-13 and IL-25. The main effector cells are eosinophils, basophils, and mast cells as well as B cells, and IL-4/IL-5 CD4 T cells. The key Th2 transcription factors are STAT6 and GATA3.[24] IL-4 is the positive feedback cytokine for Th2 cells differentiation. Besides, IL-4 stimulates B-cells to produce IgE antibodies, which in turn stimulate mast cells to release histamine, serotonin, and leukotriene to cause broncho-constriction, intestinal peristalsis, gastric fluid acidification to expel helminths. IL-5 from CD4 T cells will activate eosinophils to attack helminths. IL-10 suppresses Th1 cells differentiation and function of dendritic cells. Th2 overactivation against antigen will cause Type I hypersensitivity which is an allergic reaction mediated by IgE. Allergic rhinitis, atopic dermatitis, and asthma belong to this category of overactivation .[23] In addition to expressing different cytokines, Th2 cells also differ from Th1 cells in their cell surface glycans (oligosaccharides), which makes them less susceptible to some inducers of cell death.[25][26]
| Type 1/ Th1 | Type 2/ Th2[23] | |
|---|---|---|
| Main partner cell type | Macrophage, CD8+ T cell | B-cell, eosinophil, mast cell |
| Cytokines produced | interferon-γ and TNF-β. Interleukin-2 interleukin-10 production has been reported in activated Th1 cell.[28] | interleukin-4, interleukin-5, interleukin-6, interleukin-9, interleukin-10, interleukin-13 |
| Immune stimulation promoted | Cellular immune system. Maximizes the killing efficacy of the macrophages and the proliferation of cytotoxic CD8+ T cells. Also promotes the production of IgG, an opsonizing antibody. | Humoral immune system. Stimulates B-cells into proliferation, to induce B-cell antibody class switching, and to increase neutralizing antibody production (IgG, IgM and IgA as well as IgE antibodies). |
| Other functions | The Type 1 cytokine IFN-γ increases the production of interleukin-12 by dendritic cells and macrophages, and via positive feedback, IL-12 stimulates the production of IFN-γ in helper T cells, thereby promoting the Th1 profile. IFN-gamma also inhibits the production of cytokines such as interleukin-4, an important cytokine associated with the Type 2 response, and thus it also acts to preserve its own response. | The Type 2 response promotes its own profile using two different cytokines. Interleukin-4 acts on helper T cells to promote the production of Th2 cytokines (including itself; it is auto-regulatory), while interleukin-10 (IL-10) inhibits a variety of cytokines including interleukin-2 and IFN-γ in helper T cells and IL-12 in dendritic cells and macrophages. The combined action of these two cytokines suggests that once the T cell has decided to produce these cytokines, that decision is preserved (and also encourages other T cells to do the same). |
While we know about the types of cytokine patterns helper T cells tend to produce, we understand less about how the patterns themselves are decided. Various evidence suggests that the type of APC presenting the antigen to the T cell has a major influence on its profile. Other evidence suggests that the concentration of antigen presented to the T cell during primary activation influences its choice. The presence of some cytokines (such as the ones mentioned above) will also influence the response that will eventually be generated, but our understanding is nowhere near complete.
Th17 helper cells
Th17 helper cells are a subset of T helper cells developmentally distinct from Th1 and Th2 lineages producing interleukin 17 (IL-17). Th cells produce IL-17 which is a pro inflammatory substance. This means it is especially good at fighting extracellular pathogens and fungi.
THαβ helper cells
THαβ helper cells provide the host immunity against viruses. Their differentiation is triggered by IFN α/β or IL-10. Their key effector cytokine is IL-10. Their main effector cells are NK cells as well as CD8 T cells, IgG B cells, and IL-10 CD4 T cells. The key THαβ transcription factors are STAT1 and STAT3 as well as IRFs. IL-10 from CD4 T cells activate NK cells' ADCC to apoptose virus-infected cells and to induce host as well as viral DNA fragmentation. IFN alpha/beta can suppress transcription to avoid virus replication and transmission. Overactivation of THαβ against autoantigen will cause type 2 antibody-dependent cytotoxic hypersensitivity. Myasthenia gravis or Graves' disease belong to this category.[29]
Limitations to the Th1/Th2 model
The interactions between cytokines from the Th1/Th2 model can be more complicated in some animals. For example, the Th2 cytokine IL-10 inhibits cytokine production of both Th subsets in humans. Human IL-10 (hIL-10) suppresses the proliferation and cytokine production of all T cells and the activity of macrophages, but continues to stimulate plasma cells, ensuring that antibody production still occurs. As such, hIL-10 is not believed to truly promote the Th2 response in humans, but acts to prevent over-stimulation of helper T cells while still maximising the production of antibodies.
There are also other types of T cells that can influence the expression and activation of helper T cells, such as natural regulatory T cells, along with less common cytokine profiles such as the Th3 subset of helper T cells. Terms such as "regulatory" and "suppression" have become ambiguous after the discovery that helper CD4+ T cells are also capable of regulating (and suppressing) their own responses outside of dedicated regulatory T cells.
One major difference between regulatory T cells and effector T cells is that regulatory T cells typically serve to modulate and deactivate the immune response, while effector T cell groups usually begin with immune-promoting cytokines and then switch to inhibitory cytokines later in their life cycle. The latter is a feature of Th3 cells, which transform into a regulatory subset after its initial activation and cytokine production.
Both regulatory T cells and Th3 cells produce the cytokine transforming growth factor-beta (TGF-β) and IL-10. Both cytokines are inhibitory to helper T cells; TGF-β suppresses the activity of most of the immune system. There is evidence to suggest that TGF-β may not suppress activated Th2 cells as effectively as it might suppress naive cells, but it is not typically considered a Th2 cytokine.
The characterisation of another novel T helper subtype, T helper 17 cells (Th17)[30] has cast further doubt on the basic Th1/Th2 model. These IL-17 producing cells were initially described as a pathogenic population implicated in autoimmunity but are now thought to have their own distinct effector and regulatory functions. Of note, recent evidence suggest that functional plasticity is an intrinsic capacity of T helper cells. Indeed, a study in mice demonstrated that Th17 cells transform into Th1 cells in vivo.[31] A subsequent study furthermore showed that extensive T helper cell plasticity is also prominent in man.[32]
Many of the cytokines in this article are also expressed by other immune cells (see individual cytokines for details), and it is becoming clear that while the original Th1/Th2 model is enlightening and gives insight into the functions of helper T cells, it is far too simple to define its entire role or actions. Some immunologists question the model completely, as some in vivo studies suggest that individual helper T cells usually do not match the specific cytokine profiles of the Th model, and many cells express cytokines from both profiles.[33] That said, the Th model has still played an important part in developing our understanding of the roles and behaviour of helper T cells and the cytokines they produce during an immune response.
Recent scientific studies by Stockinger et al. revealed that another T helper subset may exist. Th9 cells are claimed to be an IL9 (interleukin 9)–producing T cell subset focused on defending helminth infections.[34]
Memory T cell
Historically, memory T cells were thought to belong to either the effector or central memory subtypes, each with their own distinguishing set of cell surface markers.[35] Central memory T cells reside in the lymph nodes while effector memory T cells lack the C-C chemokine receptor type 7 (CCR7) and L-selectin (CD62L) receptors, which prevents them from trafficking to the lymph nodes.
Additional populations of memory T cells are now known to exist. These include tissue-resident memory T (Trm) cells and virtual memory T cells.[36] The single unifying theme for all memory T cell subtypes is that they are long-lived and can expand quickly to large numbers of effector T cells upon encountering their cognate antigen. By this mechanism they provide the immune system with "memory" against previously encountered pathogens.
Role in disease
Considering the diverse and important role helper T cells play in the immune system, it is not surprising that these cells often influence the immune response against disease. They also appear to make occasional mistakes, or generate responses that would be politely considered non-beneficial. In the worst-case scenario, the helper T cell response could lead to a disaster and the death of the host. Fortunately this is a very rare occurrence.
Antitumor immunity
Hypersensitivity
The immune system must achieve a balance of sensitivity in order to respond to foreign antigens without responding to the antigens of the host itself. When the immune system responds to very low levels of antigen that it usually shouldn't respond to, a hypersensitivity response occurs. Hypersensitivity is believed to be the cause of allergy and some auto-immune disease.
Hypersensitivity reactions can be divided into four types:
- Type 1 hypersensitivity includes common immune disorders such as asthma, allergic rhinitis (hay fever), eczema, urticaria (hives) and anaphylaxis. These reactions all involve IgE antibodies, which require a Th2 response during helper T cell development. Preventive treatments, such as corticosteroids and montelukast, focus on suppressing mast cells or other allergic cells; T cells do not play a primary role during the actual inflammatory response. It's important to note that the numeral allocation of hypersensitivity "types" does not correlate (and is completely unrelated) to the "response" in the Th model.
- Type 2 and Type 3 hypersensitivity both involve complications from auto-immune or low affinity antibodies. In both of these reactions, T cells may play an accomplice role in generating these auto-specific antibodies, although some of these reactions under Type 2 hypersensitivity would be considered normal in a healthy immune system (for example, Rhesus factor reactions during child-birth is a normal immune response against child antigens). The understanding of the role of helper T cells in these responses is limited but it is generally thought that Th2 cytokines would promote such disorders. For example, studies have suggested that lupus (SLE) and other auto-immune diseases of similar nature can be linked to the production of Th2 cytokines.
- Type 4 hypersensitivity, also known as delayed type hypersensitivity, are caused via the over-stimulation of immune cells, commonly lymphocytes and macrophages, resulting in chronic inflammation and cytokine release. Antibodies do not play a direct role in this allergy type. T cells play an important role in this hypersensitivity, as they activate against the stimulus itself and promote the activation of other cells; particularly macrophages via Th1 cytokines.
Other cellular hypersensitivities include cytotoxic T cell mediated auto-immune disease, and a similar phenomenon; transplant rejection. Helper T cells are required to fuel the development of these diseases. In order to create sufficient auto-reactive killer T cells, interleukin-2 must be produced, and this is supplied by CD4+ T cells. CD4+ T cells can also stimulate cells such as natural killer cells and macrophages via cytokines such as interferon-gamma, encouraging these cytotoxic cells to kill host cells in certain circumstances.
The mechanism that killer T cells use during auto-immunity is almost identical to their response against viruses, and some viruses have been accused of causing auto-immune diseases such as Type 1 diabetes mellitus. Cellular auto-immune disease occurs because the host antigen recognition systems fail, and the immune system believes, by mistake, that a host antigen is foreign. As a result, the CD8+ T cells treat the host cell presenting that antigen as infected, and go on to destroy all host cells (or in the case of transplant rejection, transplant organ) that express that antigen.
Some of this section is a simplification. Many auto-immune diseases are more complex. A well-known example is rheumatoid arthritis, where both antibodies and immune cells are known to play a role in the pathology. Generally the immunology of most auto-immune diseases is not well understood.
HIV infection
Perhaps the best example of the importance of CD4+ T cells is demonstrated with human immunodeficiency virus (HIV) infection. HIV mainly targets lymphoid CD4+ T cells, but can infect other cells that express CD4 such as macrophages and dendritic cells (both groups express CD4 at low levels).
It has been proposed that during the non-symptomatic phase of HIV infection, the virus has a relatively low affinity towards T cells (and has a higher affinity for macrophages), resulting in a slow kill rate of CD4+ T cells by the immune system. This is initially compensated for via the production of new helper T cells from the thymus (originally from the bone marrow). Once the virus becomes lymphotropic (or T-tropic) however, it begins to infect CD4+ T cells far more efficiently (likely due to a change in the co-receptors it binds to during infection), and the immune system is overwhelmed. Of note, recent studies suggest that only ~5% of the lymphoid-derived CD4 T cells targeted by HIV are permissive and become productively infected with the virus. More than 95% of the CD4 T cells that die are resting and are unable to support productive infection. These cells undergo abortive infection with HIV.[37] Cell death is triggered when the host cell detects HIV foreign DNA intermediates and initiates a suicidal death pathway in an attempt to protect the host, leading to caspase-1 activation in inflammasomes, thus causing pyroptosis (a highly inflammatory form of programmed cell death).[38][39]
At this point chronic inflammation ensues, and functional CD4+ T cell levels begin to decrease, eventually to a point where the CD4+ T cell population is too small to recognize the full range of antigens that could potentially be detected. The depletion of CD4 T cells and the development of chronic inflammation are signature processes in HIV pathogenesis that propel progression to acquired immune deficiency syndrome (AIDS). CD4 T cell depleted to the cell count of less than 200cell/μL in blood during AIDS allows various pathogens to escape T cell recognition, thus allowing opportunistic infections that would normally elicit a helper T cell response to bypass the immune system.[40] While these complete bypass situations only occur when the helper T cell response is absolutely necessary for infection clearance, most infections increase in severity and/or duration because the immune system's helper T cells provide less efficient immune response.
Two components of the immune system are particularly affected in AIDS, due to its CD4+ T cell dependency:
- CD8+ T cells are not stimulated as effectively during the AIDS stage of HIV infection, making AIDS patients very susceptible to most viruses, including HIV itself. This decline in killing of CD4+ T cells results in the virus being produced for a longer period (the infected CD4+ T cells are not killed as quickly), increasing the proliferation of the virus, and accelerating the development of the disease.
- Antibody class switching declines significantly once helper T cell function fails. The immune system loses its ability to improve the affinity of their antibodies, and are unable to generate B cells that can produce antibody groups such as IgG and IgA. These effects are primarily due to the loss of any helper T cell that can interact with the B lymphocyte correctly. Another symptom of AIDS is the reduction in antibody levels due to a decrease in Th2 cytokines (and less interactions by helper T cells). All of these complications result in an increased susceptibility to aggressive bacterial infections, especially in areas of the body not accessible by IgM antibodies.
If the patient does not respond to (or does not receive) HIV treatment they will succumb usually to either cancers or infections; the immune system finally reaches a point where it is no longer coordinated or stimulated enough to deal with the disease.
Inhibition of CD4 T-cell expansion during HIV infection may occur due to microbial translocation in an IL-10-dependent way. Triggering PD-1 expressed on activated monocytes by its ligand PD-L1, induces IL-10 production which inhibits CD4 T-cell function.[41]
COVID-19
In coronavirus disease 2019 (COVID-19) B cell, natural killer cell, and total lymphocyte counts decline, but both CD4+ and CD8+ cells decline to a far greater extent.[42] Low CD4+ predicted greater likelihood of intensive care unit admission, and CD4+ cell count was the only parameter that predicted length of time for viral RNA clearance.[42] Despite the reduced levels of CD4+, COVID-19 patients with severe disease had higher levels of Th1 CD4+ cells than patients with moderate disease.[43]
See also
References
- Lederman S, Yellin MJ, Krichevsky A, Belko J, Lee JJ, Chess L (April 1992). "Identification of a novel surface protein on activated CD4+ T cells that induces contact-dependent B cell differentiation (help)". The Journal of Experimental Medicine. 175 (4): 1091–101. doi:10.1084/jem.175.4.1091. PMC 2119166. PMID 1348081.
- Lederman S, Yellin MJ, Inghirami G, Lee JJ, Knowles DM, Chess L (December 1992). "Molecular interactions mediating T-B lymphocyte collaboration in human lymphoid follicles. Roles of T cell-B-cell-activating molecule (5c8 antigen) and CD40 in contact-dependent help". Journal of Immunology. 149 (12): 3817–26. PMID 1281189.
- Lederman S, Yellin MJ, Cleary AM, Pernis A, Inghirami G, Cohn LE, Covey LR, Lee JJ, Rothman P, Chess L (March 1994). "T-BAM/CD40-L on helper T lymphocytes augments lymphokine-induced B cell Ig isotype switch recombination and rescues B cells from programmed cell death". Journal of Immunology. 152 (5): 2163–71. PMID 7907632.
- "Entrez Gene: CD40LG CD40 ligand (TNF superfamily, member 5, hyper-IgM syndrome)".
- Sullivan, Kathleen E. (Professor of pediatrics) Stiehm, E. Richard, 1933- (2020). Stiehm's immune deficiencies inborn errors of immunity. Academic Press. ISBN 978-0-12-817295-7. OCLC 1156337055.CS1 maint: multiple names: authors list (link)
- Oestreich, Kenneth J.; Weinmann, Amy S. (2012-10-12). "Master regulators or lineage-specifying? Changing views on CD4+ T cell transcription factors". Nature Reviews Immunology. 12 (11): 799–804. doi:10.1038/nri3321. ISSN 1474-1733. PMC 3584691.
- Ma, Cindy S. Chew, Gary Y.J. Simpson, Nicholas Priyadarshi, Archana Wong, Melanie Grimbacher, Bodo Fulcher, David A. Tangye, Stuart G. Cook, Matthew C. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. The Rockefeller University Press. OCLC 679066657.CS1 maint: multiple names: authors list (link)
- Ma, Cindy S.; Avery, Danielle T.; Chan, Anna; Batten, Marcel; Bustamante, Jacinta; Boisson-Dupuis, Stephanie; Arkwright, Peter D.; Kreins, Alexandra Y.; Averbuch, Diana; Engelhard, Dan; Magdorf, Klaus (2012-04-26). "Functional STAT3 deficiency compromises the generation of human T follicular helper cells". Blood. 119 (17): 3997–4008. doi:10.1182/blood-2011-11-392985. ISSN 0006-4971.
- Fink, Pamela J. (2013-03-21). "The Biology of Recent Thymic Emigrants". Annual Review of Immunology. 31 (1): 31–50. doi:10.1146/annurev-immunol-032712-100010. ISSN 0732-0582.
- van den Broek T, Borghans JA, van Wijk F (March 2018). "The full spectrum of human naive T cells". Nature Reviews. Immunology. 18 (6): 363–373. doi:10.1038/s41577-018-0001-y. PMID 29520044.
- van den Broek T, Delemarre EM, Janssen WJ, Nievelstein RA, Broen JC, Tesselaar K, Borghans JA, Nieuwenhuis EE, Prakken BJ, Mokry M, Jansen NJ, van Wijk F (March 2016). "Neonatal thymectomy reveals differentiation and plasticity within human naive T cells". The Journal of Clinical Investigation. 126 (3): 1126–36. doi:10.1172/JCI84997. PMC 4767338. PMID 26901814.
- Roche, Paul A.; Furuta, Kazuyuki (2015-02-27). "The ins and outs of MHC class II-mediated antigen processing and presentation". Nature Reviews Immunology. 15 (4): 203–216. doi:10.1038/nri3818. ISSN 1474-1733. PMC 6314495.
- Unanue, Emil R.; Turk, Vito; Neefjes, Jacques (2016-05-11). "Variations in MHC Class II Antigen Processing and Presentation in Health and Disease". Annual Review of Immunology. 34 (1): 265–297. doi:10.1146/annurev-immunol-041015-055420. ISSN 0732-0582.
- Wieczorek, Marek; Abualrous, Esam T.; Sticht, Jana; Álvaro-Benito, Miguel; Stolzenberg, Sebastian; Noé, Frank; Freund, Christian (2017-03-17). "Major Histocompatibility Complex (MHC) Class I and MHC Class II Proteins: Conformational Plasticity in Antigen Presentation". Frontiers in Immunology. 8. doi:10.3389/fimmu.2017.00292. ISSN 1664-3224. PMC 5355494. PMID 28367149.
- Trolle, Thomas; McMurtrey, Curtis P.; Sidney, John; Bardet, Wilfried; Osborn, Sean C.; Kaever, Thomas; Sette, Alessandro; Hildebrand, William H.; Nielsen, Morten; Peters, Bjoern (2016-02-15). "The Length Distribution of Class I–Restricted T Cell Epitopes Is Determined by Both Peptide Supply and MHC Allele–Specific Binding Preference". The Journal of Immunology. 196 (4): 1480–1487. doi:10.4049/jimmunol.1501721. ISSN 0022-1767. PMC 4744552. PMID 26783342.
- Murphy, Kenneth. (2017). Janeway's immunobiology. Garland Science. ISBN 978-0-8153-4551-0. OCLC 1020120603.
- Guy, Bruno (July 2007). "The perfect mix: recent progress in adjuvant research". Nature Reviews Microbiology. 5 (7): 396–397. doi:10.1038/nrmicro1681. ISSN 1740-1534.
- Hammer, Gianna Elena; Ma, Averil (2013-03-21). "Molecular Control of Steady-State Dendritic Cell Maturation and Immune Homeostasis". Annual Review of Immunology. 31 (1): 743–791. doi:10.1146/annurev-immunol-020711-074929. ISSN 0732-0582. PMC 4091962.
- Zamoyska, Rose (September 2007). "Why Is There so Much CD45 on T Cells?". Immunity. 27 (3): 421–423. doi:10.1016/j.immuni.2007.08.009. ISSN 1074-7613.
- Courtney, Adam H.; Shvets, Alexey A.; Lu, Wen; Griffante, Gloria; Mollenauer, Marianne; Horkova, Veronika; Lo, Wan-Lin; Yu, Steven; Stepanek, Ondrej; Chakraborty, Arup K.; Weiss, Arthur (2019-10-22). "CD45 functions as a signaling gatekeeper in T cells". Science Signaling. 12 (604): eaaw8151. doi:10.1126/scisignal.aaw8151. ISSN 1945-0877. PMC 6948007. PMID 31641081.
- Armitage RJ, Fanslow WC, Strockbine L, Sato TA, Clifford KN, Macduff BM, Anderson DM, Gimpel SD, Davis-Smith T, Maliszewski CR, et al. (May 1992). "Molecular and biological characterization of a murine ligand for CD40". Nature. 357 (6373): 80–2. doi:10.1038/357080a0. PMID 1374165.
- Noelle RJ, Roy M, Shepherd DM, Stamenkovic I, Ledbetter JA, Aruffo A (July 1992). "A 39-kDa protein on activated helper T cells binds CD40 and transduces the signal for cognate activation of B cells". Proc Natl Acad Sci U S A. 89 (14): 6550–4. doi:10.1073/pnas.89.14.6550. PMC 49539. PMID 1378631.
- Zhu J, Paul WE (September 2008). "CD4 T cells: fates, functions, and faults". Blood. 112 (5): 1557–69. doi:10.1182/blood-2008-05-078154. PMC 2518872. PMID 18725574.
- Wan YY (June 2014). "GATA3: a master of many trades in immune regulation". Trends in Immunology. 35 (6): 233–42. doi:10.1016/j.it.2014.04.002. PMC 4045638. PMID 24786134.
- Maverakis E, Kim K, Shimoda M, Gershwin ME, Patel F, Wilken R, Raychaudhuri S, Ruhaak LR, Lebrilla CB (February 2015). "Glycans in the immune system and The Altered Glycan Theory of Autoimmunity: a critical review". Journal of Autoimmunity. 57 (6): 1–13. doi:10.1016/j.jaut.2014.12.002. PMC 4340844. PMID 25578468.
- Toscano MA, Bianco GA, Ilarregui JM, Croci DO, Correale J, Hernandez JD, Zwirner NW, Poirier F, Riley EM, Baum LG, Rabinovich GA (2007). "Differential glycosylation of TH1, TH2 and TH-17 effector cells selectively regulates susceptibility to cell death". Nat Immunol. 8 (8): 825–34. doi:10.1038/ni1482. PMID 17589510.
- Rang HP, Dale MM, Riter JM, Moore PK (2003). Pharmacology. Edinburgh: Churchill Livingstone. ISBN 978-0-443-07145-4. Page 223
- Saraiva M, Christensen JR, Veldhoen M, Murphy TL, Murphy KM, O'Garra A (August 2009). "Interleukin-10 production by Th1 cells requires interleukin-12-induced STAT4 transcription factor and ERK MAP kinase activation by high antigen dose". Immunity. 31 (2): 209–19. doi:10.1016/j.immuni.2009.05.012. PMC 2791889. PMID 19646904.
- Hu W (2007). Microarray analysis of PBMC gene expression profiles after Plasmodium falciparum malarial infection (Ph.D. thesis). Johns Hopkins University.
- Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT (November 2005). "Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages". Nature Immunology. 6 (11): 1123–32. doi:10.1038/ni1254. PMID 16200070.
- Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, Ahlfors H, Wilhelm C, Tolaini M, Menzel U, Garefalaki A, Potocnik AJ, Stockinger B (March 2011). "Fate mapping of IL-17-producing T cells in inflammatory responses". Nature Immunology. 12 (3): 255–63. doi:10.1038/ni.1993. PMC 3040235. PMID 21278737.
- Larsen M, Arnaud L, Hié M, Parizot C, Dorgham K, Shoukry M, Kemula M, Barete S, Derai D, Sauce D, Amoura Z, Pène J, Yssel H, Gorochov G (September 2011). "Multiparameter grouping delineates heterogeneous populations of human IL-17 and/or IL-22 T-cell producers that share antigen specificities with other T-cell subsets". European Journal of Immunology. UPMC Paris 06 Institut National de la Santé et de la Recherche Médicale (Inserm) UMR-S 945. 41 (9): 2596–605. doi:10.1002/eji.201041131. PMID 21688259.
- Nakayamada S, Takahashi H, Kanno Y, O'Shea JJ (June 2012). "Helper T cell diversity and plasticity". Current Opinion in Immunology. 24 (3): 297–302. doi:10.1016/j.coi.2012.01.014. PMC 3383341. PMID 22341735.
- (Veldhoen and Stockinger, Dardalhon and Kuchroo both papers from Nature Immunology 2008)
- Sallusto F, Lenig D, Förster R, Lipp M, Lanzavecchia A (1999). "Two subsets of memory T lymphocytes with distinct homing potentials and effector functions". Nature. 401 (6754): 708–712. doi:10.1038/44385. PMID 10537110.
- Marusina AI, Ono Y, Merleev AA, Shimoda M, Ogawa H, Wang EA, Kondo K, Olney L, Luxardi G, Miyamura Y, Yilma TD, Villalobos IB, Bergstrom JW, Kronenberg DG, Soulika AM, Adamopoulos IE, Maverakis E (2017). "CD4+ virtual memory: Antigen-inexperienced T cells reside in the naïve, regulatory, and memory T cell compartments at similar frequencies, implications for autoimmunity". Journal of Autoimmunity. 77 (2): 76–88. doi:10.1016/j.jaut.2016.11.001. PMC 6066671. PMID 27894837.
- Doitsh G, Cavrois M, Lassen KG, Zepeda O, Yang Z, Santiago ML, et al. (November 2010). "Abortive HIV infection mediates CD4 T cell depletion and inflammation in human lymphoid tissue". Cell. 143 (5): 789–801. doi:10.1016/j.cell.2010.11.001. PMC 3026834. PMID 21111238.
- Doitsh G, Galloway NL, Geng X, Yang Z, Monroe KM, Zepeda O, et al. (January 2014). "Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection". Nature. 505 (7484): 509–14. doi:10.1038/nature12940. PMC 4047036. PMID 24356306.
- Monroe KM, Yang Z, Johnson JR, Geng X, Doitsh G, Krogan NJ, Greene WC (January 2014). "IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV". Science. 343 (6169): 428–32. doi:10.1126/science.1243640. PMC 3976200. PMID 24356113.
- "CD4 Count". www.aids.gov. Retrieved 2015-04-30.
- Said EA, Dupuy FP, Trautmann L, Zhang Y, Shi Y, El-Far M, et al. (April 2010). "Programmed death-1-induced interleukin-10 production by monocytes impairs CD4+ T cell activation during HIV infection". Nature Medicine. 16 (4): 452–9. doi:10.1038/nm.2106. PMC 4229134. PMID 20208540.
- Huang W, Berube J, McNamara M, Saksena S, O'Gorman M (2020). "Lymphocyte Subset Counts in COVID-19 Patients: A Meta-Analysis". Cytometry Part A. 97 (8): 772–776. doi:10.1002/cyto.a.24172. PMC 7323417. PMID 32542842.
- Perlman S (2020). "COVID-19 poses a riddle for the immune system". Nature. 584 (782): 345–346. doi:10.1038/d41586-020-02379-1. PMID 32807916.
Further reading
- Doitsh G, Greene WC (2016). "Dissecting How CD4 T Cells Are Lost During HIV Infection". Cell Host Microbe. 19 (3): 280–91. doi:10.1016/j.chom.2016.02.012. PMC 4835240. PMID 26962940.
- Kanno Y, Vahedi G, Hirahara K, Singleton K, O'Shea JJ (2012). "Transcriptional and Epigenetic Control of T Helper Cell Specification: Molecular Mechanisms Underlying Commitment and Plasticity". Annual Review of Immunology. 30: 707–731. doi:10.1146/annurev-immunol-020711-075058. PMC 3314163. PMID 22224760.
External links
| Wikimedia Commons has media related to T helper cell. |
- "T-cell Group". T-Cells. Cardiff University.