Hydrogenase
A hydrogenase is an enzyme that catalyses the reversible oxidation of molecular hydrogen (H2), as shown below:
-
H2 + Aox → 2H+ + Ared
(1)
-
2H+ + Dred → H2 + Dox
(2)
Hydrogen uptake (1) is coupled to the reduction of electron acceptors such as oxygen, nitrate, sulfate, carbon dioxide (CO
2), and fumarate. On the other hand, proton reduction (2) is coupled to the oxidation of electron donors such as ferredoxin (FNR), and serves to dispose excess electrons in cells (essential in pyruvate fermentation). Both low-molecular weight compounds and proteins such as FNRs, cytochrome c3, and cytochrome c6 can act as physiological electron donors or acceptors for hydrogenases.[1]
Structural classification
It has been estimated that 99% of all organisms utilize dihydrogen, H2. Most of these species are microbes and their ability to use H2 as a metabolite arises from the expression of H2 metalloenzymes known as hydrogenases.[2] Hydrogenases are sub-classified into three different types based on the active site metal content: iron-iron hydrogenase, nickel-iron hydrogenase, and iron hydrogenase.
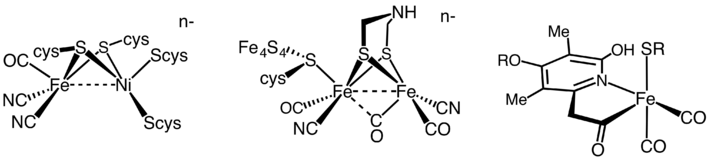
All hydrogenases catalyze reversible H2 uptake, but while the [FeFe] and [NiFe] hydrogenases are true redox catalysts, driving H2 oxidation and proton (H+) reduction (equation 3), the [Fe] hydrogenases catalyze the reversible heterolytic cleavage of H2 shown by reaction (4).
-
H2 ⇌ 2 H+ + 2 e−
(3)
-
H2 ⇌ H+ + H−
(4)
Until 2004, the [Fe]-only hydrogenase was believed to be "metal-free". Then, Thauer et al. showed that the metal-free hydrogenases in fact contain iron atom in its active site. As a result, those enzymes previously classified as "metal-free" are now named [Fe]-only hydrogenases. This protein contains only a mononuclear Fe active site and no iron-sulfur clusters, in contrast to the [FeFe] hydrogenases. [NiFe] and [FeFe] hydrogenases have some common features in their structures: Each enzyme has an active site and a few Fe-S clusters that are buried in protein. The active site, which is believed to be the place where catalysis takes place, is also a metallocluster, and each metal is coordinated by carbon monoxide (CO) and cyanide (CN−) ligands.[3]
[NiFe] hydrogenase
The [NiFe] hydrogenases are heterodimeric proteins consisting of small (S) and large (L) subunits. The small subunit contains three iron-sulfur clusters while the large subunit contains the active site, a nickel-iron centre which is connected to the solvent by a molecular tunnel.[4][5] In some [NiFe] hydrogenases, one of the Ni-bound cysteine residues is replaced by selenocysteine. On the basis of sequence similarity, however, the [NiFe] and [NiFeSe] hydrogenases should be considered a single superfamily. To date, periplasmic, cytoplasmic, and cytoplasmic membrane-bound hydrogenases have been found. The [NiFe] hydrogenases, when isolated, are found to catalyse both H2 evolution and uptake, with low-potential multihaem cytochromes such as cytochrome c3 acting as either electron donors or acceptors, depending on their oxidation state.[4] Generally speaking, however, [NiFe] hydrogenases are more active in oxidizing H2. A wide spectrum of H2 affinities have also been observed in H2-oxidizing hydrogenases.[6]
Like [FeFe] hydrogenases, [NiFe] hydrogenases are known to be usually deactivated by molecular oxygen (O2). Hydrogenase from Ralstonia eutropha, and several other so-called Knallgas-bacteria, were found to be oxygen-tolerant.[4][7] The soluble [NiFe] hydrogenase from Ralstonia eutropha H16 be conveniently produced on heterotrophic growth media.[8][9] This finding increased hope that hydrogenases can be used in photosynthetic production of molecular hydrogen via splitting water.
[FeFe] hydrogenase
The hydrogenases containing a di-iron center with a bridging dithiolate cofactor are called [FeFe] hydrogenases.[10] Three families of [FeFe] hydrogenases are recognized:
- cytoplasmic, soluble, monomeric hydrogenases, found in strict anaerobes such as Clostridium pasteurianum and Megasphaera elsdenii. They catalyse both H2 evolution and uptake.
- periplasmic, heterodimeric hydrogenases from Desulfovibrio spp., which can be purified aerobically.
- soluble, monomeric hydrogenases, found in chloroplasts of green alga Scenedesmus obliquus, catalyses H2 evolution. The [Fe2S2] ferredoxin functions as natural electron donor linking the enzyme to the photosynthetic electron transport chain.
In contrast to [NiFe] hydrogenases, [FeFe] hydrogenases are generally more active in production of molecular hydrogen. Turnover frequency (TOF) in the order of 10,000 s−1 have been reported in literature for [FeFe] hydrogenases from Clostridium pasteurianum.[11] This has led to intense research focusing on use of [FeFe] hydrogenase for sustainable production of H2.[12]
The active site of the diiron hydrogenase is known as the H-cluster. The H-cluster consists of a [4Fe4S] cubane shaped structure, coupled to the low valent diiron co-factor by a cysteine derived thiol. The diiron co-factor includes two iron atoms, connected by a bridging aza-dithiolate ligand (-SCH2-NH-CH2S-, adt), the iron atoms are coordinated by carbonyl and cyanide ligands.[13]
[FeFe]-hydrogenases can be separated into four distinct phylogenetic groups A−D.[14] Group A consists of prototypical and bifurcating [FeFe]-hydrogenases. In nature, prototypical [FeFe]-hydrogenases perform hydrogen turnover using ferredoxin as a redox partner while bifurcating types perform the same reaction using both ferredoxin and NAD(H) as electron donor or acceptor.[15] In order to conserve energy, anaerobic bacteria use electron bifurcation where exergonic and endergonic redox reactions are coupled to circumvent thermodynamic barriers. Group A comprises the best characterized and catalytically most active enzymes such as the [FeFe]-hydrogenase from Chlamydomonas reinhardtii (CrHydA1),[16] Desulfovibrio desulfuricans (DdHydAB or DdH),[17] and Clostridium pasteurianum and Clostridium acetobutylicum (CpHydA1 and CaHydA1, referred to as CpI and CaI).[18] No representative examples of Group B has been characterized yet but it is phylogenetically distinct even when it shares similar amino acid motifs around the H-cluster as Group A [FeFe]-hydrogenases. Group C has been classified as “sensory” based on the presence of a Per-Arnt-Sim domain.[19][20] One example of a Group C [FeFe]-hydrogenase is from Thermotoga maritima (TmHydS) which shows only modest catalytic rates compared to Group A enzymes and an apparent high sensitivity toward hydrogen (H2).[21] A closely related subclass from Group D has a similar location on the bacterial gene and share similar domain structure to a subclass from Group E but it lacks the PAS domain.[14][19]
[Fe]-only hydrogenase
5,10-methenyltetrahydromethanopterin hydrogenase (EC 1.12.98.2) found in methanogenic Archaea contains neither nickel nor iron-sulfur clusters but an iron-containing cofactor that was recently characterized by X-ray diffraction.[22]
Unlike the other two types, [Fe]-only hydrogenases are found only in some hydrogenotrophic methanogenic archaea. They also feature a fundamentally different enzymatic mechanism in terms of redox partners and how electrons are delivered to the active site. In [NiFe] and [FeFe] hydrogenases, electrons travel through a series of metallorganic clusters that comprise a long distance; the active site structures remain unchanged during the whole process. In [Fe]-only hydrogenases, however, electrons are directly delivered to the active site via a short distance. Methenyl-H4MPT+, a cofactor, directly accepts the hydride from H2 in the process. [Fe]-only hydrogenase is also known as H2-forming methylenetetrahydromethanopterin (methylene-H4MPT) dehydrogenase, because its function is the reversible reduction of methenyl-H4MPT+ to methylene-H4MPT.[23] The hydrogenation of a methenyl-H4MPT+ occurs instead of H2 oxidation/production, which is the case for the other two types of hydrogenases. While the exact mechanism of the catalysis is still under study, recent finding suggests that molecular hydrogen is first heterolytically cleaved by Fe(II), followed by transfer of hydride to the carbocation of the acceptor.[24]
Mechanism
The molecular mechanism by which protons are converted into hydrogen molecules within hydrogenases is still under extensive study. One popular approach employs mutagenesis to elucidate roles of amino acids and/or ligands in different steps of catalysis such as intramolecular transport of substrates. For instance, Cornish et al. conducted mutagenesis studies and found out that four amino acids located along the putative channel connecting the active site and protein surface are critical to enzymatic function of [FeFe] hydrogenase from Clostridium pasteurianum (CpI).[25] On the other hand, one can also rely on computational analysis and simulations. Nilsson Lill and Siegbahn have recently taken this approach in investigating the mechanism by which [NiFe] hydrogenases catalyze H2 cleavage.[26] The two approaches are complementary and can benefit one another. In fact, Cao and Hall combined both approaches in developing the model that describes how hydrogen molecules are oxidized or produced within the active site of [FeFe] hydrogenases.[27] While more research and experimental data are required to complete our understanding of the mechanism, these findings have allowed scientists to apply the knowledge in, e.g., building artificial catalysts mimicking active sites of hydrogenases.[28]
Biological function
Assuming that the Earth's atmosphere was initially rich in hydrogen, scientists hypothesize that hydrogenases were evolved to generate energy from/as molecular H2. Accordingly, hydrogenases can either help microorganisms to proliferate under such conditions, or to set up ecosystems empowered by H2.[29] Microbial communities driven by molecular hydrogen have, in fact, been found in deep-sea settings where other sources of energy from photosynthesis are not available. Based on these grounds, the primary role of hydrogenases are believed to be energy generation, and this can be sufficient to sustain an ecosystem.
Recent studies have revealed other biological functions of hydrogenases. To begin with, bidirectional hydrogenases can also act as "valves" to control excess reducing equivalents, especially in photosynthetic microorganisms. Such a role makes hydrogenases play a vital role in anaerobic metabolism.[30][31] Moreover, hydrogenases may also be involved in membrane-linked energy conservation through the generation of a transmembrane protonmotive force.[15]There is a possibility that hydrogenases have been responsible for bioremediation of chlorinated compounds. Hydrogenases proficient in H2 uptake can help heavy metal contaminants to be recovered in intoxicated forms. These uptake hydrogenases have been recently discovered in pathogenic bacteria and parasites and are believed to be involved in their virulence.[15]
Applications
Hydrogenases were first discovered in the 1930s,[32] and they have since attracted interest from many researchers including inorganic chemists who have synthesized a variety of hydrogenase mimics. The soluble [NiFe] hydrogenase from Ralstonia eutropha H16 is a promising candidate enzyme for H2-based biofuel application as it favours H2 oxidation and is relatively oxygen-tolerant. It can be produced on heterotrophic growth media[8] and purified via anion exchange and size exclusion chromatography matrices.[9] Understanding the catalytic mechanism of hydrogenase might help scientists design clean biological energy sources, such as algae, that produce hydrogen.[33]
Biological hydrogen production
Various systems are capable of splitting water into O2 and H+ from incident sunlight. Likewise, numerous catalysts, either chemical or biological, can reduce the produced H+ into H2. Different catalysts require unequal overpotential for this reduction reaction to take place. Hydrogenases are attractive since they require a relatively low overpotential. In fact, its catalytic activity is more effective than platinum, which is the best known catalyst for H2 evolution reaction.[34] Among three different types of hydrogenases, [FeFe] hydrogenases is considered as a strong candidate for an integral part of the solar H2 production system since they offer an additional advantage of high TOF (over 9000 s−1)[6].
Low overpotential and high catalytic activity of [FeFe] hydrogenases are accompanied by high O2 sensitivity. It is necessary to engineer them O2-tolerant for use in solar H2 production since O2 is a by-product of water splitting reaction. Past research efforts by various groups around the world have focused on understanding the mechanisms involved in O2-inactivation of hydrogenases.[5][35] For instance, Stripp et al. relied on protein film electrochemistry and discovered that O2 first converts into a reactive species at the active site of [FeFe] hydrogenases, and then damages its [4Fe-4S] domain.[36] Cohen et al. investigated how oxygen can reach the active site that is buried inside the protein body by molecular dynamics simulation approach; their results indicate that O2 diffuses through mainly two pathways that are formed by enlargement of and interconnection between cavities during dynamic motion.[37] These works, in combination with other reports, suggest that inactivation is governed by two phenomena: diffusion of O2 to the active site, and destructive modification of the active site.
Despite these findings, research is still under progress for engineering oxygen tolerance in hydrogenases. While researchers have found oxygen-tolerant [NiFe] hydrogenases, they are only efficient in hydrogen uptake and not production[21]. Bingham et al.’s recent success in engineering [FeFe] hydrogenase from clostridium pasteurianum was also limited to retained activity (during exposure to oxygen) for H2 consumption, only.[38]
Hydrogenase-based biofuel cells
Typical enzymatic biofuel cells involve the usage of enzymes as electrocatalysts at either both cathode and anode or at one electrode. In hydrogenase-based biofuel cells, hydrogenase enzymes are present at the anode for H2 oxidation.[9][4][39]
Principle
The bidirectional or reversible reaction catalyzed by hydrogenase allows for the capture and storage of renewable energy as fuel with use on demand. This can be demonstrated through the chemical storage of electricity obtained from a renewable source (e.g. solar, wind, hydrothermal) as H2 during periods of low energy demands. When energy is desired, H2 can be oxidized to produce electricity.[39]
Advantages
This is one solution to the challenge in the development of technologies for the capture and storage of renewable energy as fuel with use on demand. The generation of electricity from H2 is comparable with the similar functionality of Platinum catalysts minus the catalyst poisoning, and thus is very efficient. In the case of H2/O2 fuel cells, where the product is water, there is no production of greenhouse gases.[39]
Biochemical classification
hydrogen dehydrogenase (hydrogen:NAD+ oxidoreductase)
- H2 + NAD+ ⇌ H+ + NADH
- EC 1.12.1.3
hydrogen dehydrogenase (NADP) (hydrogen:NADPH+ oxidoreductase)
- H2 + NADP+ ⇌ H+ + NADPH
- EC 1.12.2.1
cytochrome-c3 hydrogenase (hydrogen:ferricytochrome-c3 oxidoreductase)
- 2H2 + ferricytochrome c3 ⇌ 4H+ + ferrocytochrome c3
- EC 1.12.5.1
hydrogen:quinone oxidoreductase
- H2 + menaquinone ⇌ menaquinol
- EC 1.12.7.2
ferredoxin hydrogenase (hydrogen:ferredoxin oxidoreductase)
- H2 + oxidized ferredoxin ⇌ 2H+ + reduced ferredoxin
- EC 1.12.98.1
coenzyme F420 hydrogenase (hydrogen:coenzyme F420 oxidoreductase)
- H2 + coenzyme F420 ⇌ reduced coenzyme F420
- EC 1.12.99.6
hydrogenase (acceptor) (hydrogen:acceptor oxidoreductase)
- H2 + A ⇌ AH2
- EC 1.12.98.2
5,10-methenyltetrahydromethanopterin hydrogenase (hydrogen:5,10-methenyltetrahydromethanopterin oxidoreductase)
- H2 + 5,10-methenyltetrahydromethanopterin ⇌ H+ + 5,10-methylenetetrahydromethanopterin
- EC 1.12.98.3
Methanosarcina-phenazine hydrogenase [hydrogen:2-(2,3-dihydropentaprenyloxy)phenazine oxidoreductase]
- H2 + 2-(2,3-dihydropentaprenyloxy)phenazine ⇌ 2-dihydropentaprenyloxyphenazine
References
- Vignais, P.M.; Billoud, B.; Meyer, J. (2001). "Classification and phylogeny of hydrogenases". FEMS Microbiol. Rev. 25 (4): 455–501. doi:10.1111/j.1574-6976.2001.tb00587.x. PMID 11524134.
- Lubitz, Wolfgang; Ogata, Hideaki; Rüdiger, Olaf; Reijerse, Edward (2014). "Hydrogenases". Chemical Reviews. 114 (8): 4081–148. doi:10.1021/cr4005814. PMID 24655035.
- Fontecilla-Camps, J.C.; Volbeda, A.; Cavazza, C.; Nicolet Y. (2007). "Structure/function relationships of [NiFe]- and [FeFe]-hydrogenases". Chem Rev. 107 (10): 4273–4303. doi:10.1021/cr050195z. PMID 17850165.
- Jugder, Bat-Erdene; Welch, Jeffrey; Aguey-Zinsou, Kondo-Francois; Marquis, Christopher P. (2013-05-14). "Fundamentals and electrochemical applications of [Ni–Fe]-uptake hydrogenases". RSC Advances. 3 (22): 8142. doi:10.1039/c3ra22668a. ISSN 2046-2069.
- Liebgott, P.P.; Leroux, F.; Burlat, B.; Dementin, S.; Baffert, C.; Lautier, T.; Fourmond, V.; Ceccaldi, P.; Cavazza, C.; Meynial-Salles, I.; Soucaille, P.; Fontecilla-Camps, J.C.; Guigliarelli, B.; Bertrand, P.; Rousset, M.; Léger, C. (2010). "Relating diffusion along the substrate tunnel and oxygen sensitivity in hydrogenase". Nat. Chem. Biol. 6 (1): 63–70. doi:10.1038/nchembio.276. PMID 19966788.
- Greening C, Berney M, Hards K, Cook GM, Conrad R (2014). "A soil actinobacterium scavenges atmospheric H2 using two membrane-associated, oxygen-dependent hydrogenases". Proc. Natl. Acad. Sci. U.S.A. 111 (11): 4257–61. Bibcode:2014PNAS..111.4257G. doi:10.1073/pnas.1320586111. PMC 3964045. PMID 24591586.
- Burgdorf, T.; Buhrke, T.; van der Linden, E.; Jones, A.; Albracht, S.; Friedrich, B. (2005). "[NiFe]-Hydrogenases of Ralstonia eutropha H16: Modular Enzymes for Oxygen-Tolerant Biological Hydrogen Oxidation". J. Mol. Microbiol. Biotechnol. 10 (2–4): 181–196. doi:10.1159/000091564. PMID 16645314.
- Jugder, Bat-Erdene; Chen, Zhiliang; Ping, Darren Tan Tek; Lebhar, Helene; Welch, Jeffrey; Marquis, Christopher P. (2015-03-25). "An analysis of the changes in soluble hydrogenase and global gene expression in Cupriavidus necator ( Ralstonia eutropha ) H16 grown in heterotrophic diauxic batch culture". Microbial Cell Factories. 14 (1): 42. doi:10.1186/s12934-015-0226-4. ISSN 1475-2859. PMC 4377017. PMID 25880663.
- Jugder, Bat-Erdene; Lebhar, Helene; Aguey-Zinsou, Kondo-Francois; Marquis, Christopher P. (2016-01-01). "Production and purification of a soluble hydrogenase from Ralstonia eutropha H16 for potential hydrogen fuel cell applications". MethodsX. 3: 242–250. doi:10.1016/j.mex.2016.03.005. PMC 4816682. PMID 27077052.
- Berggren, G.; Adamska, A.; Lambertz, C.; Simmons, T. R.; Esselborn, J.; Atta, A.; Gambarelli, S.; Mouesca, J.-M.; Reijerse, E.; Lubitz, W.; Happe, T.; Artero, V.; Fontecave, M. (2013). "Biomimetic assembly and activiation of [FeFe]-hydrogenases". Nature. 499 (7456): 66–69. Bibcode:2013Natur.499...66B. doi:10.1038/nature12239. PMC 3793303. PMID 23803769.
- Madden C, Vaughn MD, Díez-Pérez I, Brown KA, King PW, Gust D, Moore AL, Moore TA (January 2012). "Catalytic turnover of [FeFe]-hydrogenase based on single-molecule imaging". Journal of the American Chemical Society. 134 (3): 1577–82. doi:10.1021/ja207461t. PMID 21916466.
- Smith PR, Bingham AS, Swartz JR (2012). "Generation of hydrogen from NADPH using an [FeFe] hydrogenase". International Journal of Hydrogen Energy. 37 (3): 2977–2983. doi:10.1016/j.ijhydene.2011.03.172.
- Németh, Brigitta; Esmieu, Charlène; Redman, Holly J.; Berggren, Gustav (2019). "Monitoring H-cluster assembly using a semi-synthetic HydF protein". Dalton Transactions. 48 (18): 5978–5986. doi:10.1039/C8DT04294B. ISSN 1477-9226. PMID 30632592.
- Land, Henrik; Senger, Moritz; Berggren, Gustav; Stripp, Sven T. (2020-05-28). "Current State of [FeFe]-Hydrogenase Research: Biodiversity and Spectroscopic Investigations". ACS Catalysis. 10 (13): 7069–7086. doi:10.1021/acscatal.0c01614. ISSN 2155-5435.
- Schuchmann, Kai; Chowdhury, Nilanjan Pal; Müller, Volker (2018-12-04). "Complex Multimeric [FeFe] Hydrogenases: Biochemistry, Physiology and New Opportunities for the Hydrogen Economy". Frontiers in Microbiology. 9. doi:10.3389/fmicb.2018.02911. ISSN 1664-302X.
- HAPPE, Thomas; NABER, J. Dirk (June 1993). "Isolation, characterization and N-terminal amino acid sequence of hydrogenase from the green alga Chlamydomonas reinhardtii". European Journal of Biochemistry. 214 (2): 475–481. doi:10.1111/j.1432-1033.1993.tb17944.x. ISSN 0014-2956.
- Glick, Bernard R.; Martin, William G.; Martin, Stanley M. (1980-10-01). "Purification and properties of the periplasmic hydrogenase from Desulfovibrio desulfuricans". Canadian Journal of Microbiology. 26 (10): 1214–1223. doi:10.1139/m80-203. ISSN 0008-4166.
- Nakos, George; Mortenson, Leonard (March 1971). "Purification and properties of hydrogenase, an iron sulfur protein, from Clostridium pasteurianum W5". Biochimica et Biophysica Acta (BBA) - Enzymology. 227 (3): 576–583. doi:10.1016/0005-2744(71)90008-8. ISSN 0005-2744.
- Calusinska, Magdalena; Happe, Thomas; Joris, Bernard; Wilmotte, Annick (2010-06-01). "The surprising diversity of clostridial hydrogenases: a comparative genomic perspective". Microbiology. 156 (6): 1575–1588. doi:10.1099/mic.0.032771-0. ISSN 1350-0872.
- Greening, Chris; Biswas, Ambarish; Carere, Carlo R; Jackson, Colin J; Taylor, Matthew C; Stott, Matthew B; Cook, Gregory M; Morales, Sergio E (2015-09-25). "Genomic and metagenomic surveys of hydrogenase distribution indicate H2 is a widely utilised energy source for microbial growth and survival". The ISME Journal. 10 (3): 761–777. doi:10.1038/ismej.2015.153. ISSN 1751-7362. PMC 4817680.
- Chongdar, Nipa; Birrell, James A.; Pawlak, Krzysztof; Sommer, Constanze; Reijerse, Edward J.; Rüdiger, Olaf; Lubitz, Wolfgang; Ogata, Hideaki (2018-01-09). "Unique Spectroscopic Properties of the H-Cluster in a Putative Sensory [FeFe] Hydrogenase". Journal of the American Chemical Society. 140 (3): 1057–1068. doi:10.1021/jacs.7b11287. ISSN 0002-7863.
- Shima S, Pilak O, Vogt S, Schick M, Stagni MS, Meyer-Klaucke W, Warkentin E, Thauer RK, Ermler U (July 2008). "The crystal structure of [Fe]-hydrogenase reveals the geometry of the active site". Science. 321 (5888): 572–5. Bibcode:2008Sci...321..572S. doi:10.1126/science.1158978. PMID 18653896.
- Salomone-Stagnia, M.; Stellatob, F.; Whaleyc, C.M.; Vogtd, S.; Moranteb, S.; Shimad, S.; Rauchfuss, T.B.; Meyer-Klaucke, W.; model systems: an X-ray absorption near edge spectroscopy study (2010). "The iron-site structure of [Fe]-hydrogenase". Dalton Transactions. 39 (12): 3057–3064. doi:10.1039/b922557a. PMC 3465567. PMID 20221540.
- Hiromoto, T.; Warkentin, E.; Moll, J.; Ermler, U.; Shima, S. (2009). "Iron-Chromophore Circular Dichroism of [Fe]-Hydrogenase: The Conformational Change Required for H2 Activation". Angew. Chem. Int. Ed. 49 (51): 9917–9921. doi:10.1002/anie.201006255. PMID 21105038.
- Cornish, A.J.; Gärtner, K.; Yang, H.; Peters, J.W.; Hegg, E.L. (2011). "Mechanism of Proton Transfer in [FeFe]-Hydrogenase from Clostridium Pasteurianum". J. Biol. Chem. 286 (44): 38341–38347. doi:10.1074/jbc.M111.254664. PMC 3207428. PMID 21900241.
- Lill, S.O.N.; Siegbahn, P.E.M. (2009). "An Autocatalytic Mechanism for NiFe-Hydrogenase: Reduction to Ni(I) Followed by Oxidative Addition". Biochemistry. 48 (5): 1056–1066. doi:10.1021/bi801218n. PMID 19138102.
- Cao, Z.; Hall, M.B. (2001). "Modeling the Active Sites in Metalloenzymes. 3. Density Functional Calculations on Models for [Fe]-Hydrogenase: Structures and Vibrational Frequencies of the Observed Redox Forms and the Reaction Mechanism at the Diiron Active Center". J. Am. Chem. Soc. 123 (16): 3734–3742. doi:10.1021/ja000116v. PMID 11457105.
- Tard, C.; Liu, X.; Ibrahim, S.K.; Bruschi, M.; Gioia, L.D.; Davies, S.C.; Yang, X.; Wang, L.S.; Sawers, G.; Pickett, C.J. (2005). "Synthesis of the H-cluster framework of iron-only hydrogenase". Nature. 433 (7026): 610–613. Bibcode:2005Natur.433..610T. doi:10.1038/nature03298. PMID 15703741.
- Vignais, P.M.; Billoud, B. (2007). "Occurrence, Classification and Biological Function of Hydrogenases: An Overview". Chem. Rev. 107 (10): 4206–4272. doi:10.1021/cr050196r. PMID 17927159.
- Adams, M.W.W.; Stiefel, E.I. (1998). "Biological hydrogen production: Not so elementary". Science. 282 (5395): 1842–1843. doi:10.1126/science.282.5395.1842. PMID 9874636.
- Frey, M. (2002). "Hydrogenases: hydrogen-activating enzymes". ChemBioChem. 3 (2–3): 153–160. doi:10.1002/1439-7633(20020301)3:2/3<153::AID-CBIC153>3.0.CO;2-B. PMID 11921392.
- Thauer, R. K., "Biochemistry of methanogenesis: a tribute to Marjory Stephenson", Microbiology, 1998, 144, 2377-2406.
- Florin, L.; Tsokoglou, A.; Happe, T. (2001). "A novel type of iron hydrogenase in the green alga Scenedesmus obliquus is linked to the photosynthetic electron transport chain". J. Biol. Chem. 276 (9): 6125–6132. doi:10.1074/jbc.M008470200. PMID 11096090.
- Hinnemann, B.; Moses, P.G.; Bonde, J.; Jørgensen, K.P.; Nielsen, J.H.; Horch, S.; Chorkendorff, I.; Nørskov, J.K. (2005). "Biomimetic hydrogen evolution: MoS2 nanoparticles as catalyst for hydrogen evolution". J. Am. Chem. Soc. 127 (15): 5308–5309. doi:10.1021/ja0504690. PMID 15826154.
- Goris, T.; Wait, A.F.; Saggu, M.; Fritsch, J.; Heidary, N.; Stein, M.; Zebger, I.; Lendzian, F.; Armstrong, F.A.; Friedrich, B.; Lenz, O. (2011). "A unique iron-sulfur cluster is crucial for oxygen tolerance of a [NiFe]-hydrogenase". Nat. Chem. Biol. 7 (5): 310–318. doi:10.1038/nchembio.555. PMID 21390036.
- Stripp, S.T.; Goldet, G.; Brandmayr, C.; Sanganas, O.; Vincent, K.A.; Haumann, M.; Armstrong, F.A.; Happe, T. (2009). "How oxygen attacks [FeFe] hydrogenases from photosynthetic organisms". Proc. Natl. Acad. Sci. 106 (41): 17331–17336. Bibcode:2009PNAS..10617331S. doi:10.1073/pnas.0905343106. PMC 2765078. PMID 19805068.
- Cohen, J.; Kim, K.; King, P.; Seibert, M.; Schulten, K. (2005). "Finding gas diffusion pathways in proteins: application to O2 and H2 transport in CpI [FeFe]-hydrogenase and the role of packing defects". Structure. 13 (9): 1321–1329. doi:10.1016/j.str.2005.05.013. PMID 16154089.
- Bingham, A.S.; Smith, P.R.; Swartz, J.R. (2012). "Evolution of an [FeFe] hydrogenase with decreased oxygen sensitivity". International Journal of Hydrogen Energy. 37 (3): 2965–2976. doi:10.1016/j.ijhydene.2011.02.048.
- Lubitz, W.; Ogata, H.; Rudiger, O.; Reijerse, E. (2014). "Hydrogenases". Chem. Rev. 114 (8): 2081–4148. doi:10.1021/cr4005814. PMID 24655035.
External links
- 2B0J - PDB Structure of the Apoenzyme of the Iron-sulphur cluster-free hydrogenase from Methanothermococcus jannaschii
- 1HFE - PDB structure of [FeFe]-hydrogenase from Desulfovibrio desulfuricans
- 1C4A - PDB structure of [FeFe]-hydrogenase from Clostridium pasteurianum
- 1UBR - PDB structure of [NiFe]-hydrogenase from Desulfovibrio vulgaris
- 1CC1 - PDB structure of [NiFeSe]-hydrogenase from Desulfomicrobium baculatum
- Animation - Mechanism of [NiFe]-hydrogenase
