Mechanism of anoxic depolarization in the brain
Anoxic depolarization is a progressive and uncontrollable depolarization of neurons during stroke or brain ischemia in which there is an inadequate supply of blood to the brain.[1] Anoxic depolarization is induced by the loss of neuronal selective membrane permeability and the ion gradients across the membrane that are needed to support neuronal activity. Normally, the Na+/K+-ATPase pump maintains the transmembrane gradients of K+ and Na+ ions, but with anoxic brain injury, the supply of energy to drive this pump is lost.[2] The hallmarks of anoxic depolarization are increased concentrations of extracellular K+ ions, intracellular Na+ and Ca2+ ions, and extracellular glutamate and aspartate. Glutamate and aspartate are normally present as the brain's primary excitatory neurotransmitters, but high concentrations activate a number of downstream apoptotic and necrotic pathways. This results in neuronal dysfunction and death.[3]
Neural signal under normal oxygen uptake
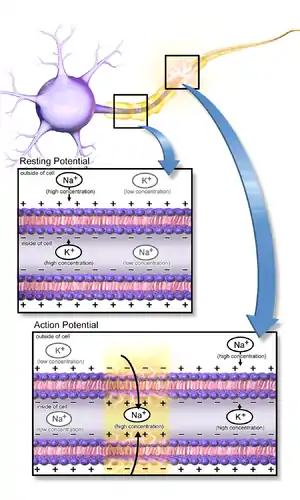
Neurons function in the central nervous system by generating signals from synapses, and this only works in the proper chemical environment.[4] An electrical signal is mediated by the sodium channels and leaky potassium channels in which intracellular K+ ion concentration is higher than its corresponding extracellular concentration, whereas extracellular concentrations of Na+, Ca2+, and Cl− ions are higher than the corresponding intracellular concentrations. This uneven distribution of ions is maintained by the Na+/K+ ATPase pump which actively pumps Na+ out and K+ into the cell in a ratio of 3:2 per ATP used. A neuron has a resting membrane potential of -70mV due to the leaky potassium channels.[5] As a neuron depolarizes due to Na+ ion influx through the sodium channels, the membrane reaches a threshold potential and then fires an all or none action potential, which either propagates down the axon or passes on to the other neurons via several gap junctions that link them.[4]
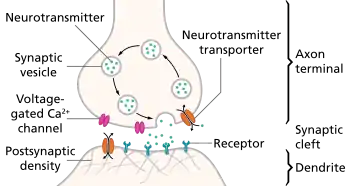
A chemical signal (synaptic transmission) begins with the action potential that propagates down the axon of so-called presynaptic terminal to trigger Ca2+ influx, which causes the synaptic vesicles to fuse and release neurotransmitters, via exocytosis, to the synaptic cleft.[5][6] Released neurotransmitters then bind their specific neuroreceptors at the postsynaptic membrane, or activate their specific ligand-gated ion channels, to fire an action potential that can be either excitatory or inhibitory, depending on the nature of the ligand-gated ion channel. Neurotransmitters are removed from the synaptic cleft by either enzymatic degradation or re-uptake by the same presynaptic neuron, via endocytosis or specific neurotransmitter transporters.[4]
Brain energy crisis
Stroke onset
Within a few seconds of stroke onset, the brain responds by entering a state of metabolic depression, in which energy consumption is reduced to compensate for the reduction in energy production. Metabolic depression occurs as a result of suppressed synaptic transmission and hyperpolarization.
The suppression of synaptic transmission occurs because the presynaptic impulse temporarily fails to trigger the release of neurotransmitters, which, coupled with the altered ion conductance and a change in postsynaptic neuroreceptors, makes synapses unresponsive to neurotransmitter binding, thereby inhibiting postsynaptic excitation.[5]
Hyperpolarization, on the other hand, is employed to reduce neuronal activity by establishing a high threshold potential for firing across an action potential. This energy-conserving response is due to the continuous inward current of K+ ions, which help maintain the membrane ion gradient until the resistance is broken and anoxic depolarization begins.[5]
Imbalance in ion-homeostasis
Maintaining a balance between the intracellular and extracellular ionic concentrations at the postsynaptic terminal is critical to normal neuronal function. During oxygen depletion to the brain, two events that initiate, as well as propagate, anoxic depolarization involve excessive cationic influxes, as well as the outflow of ATP, at the postsynaptic terminal.[1] The receptors that allow this influx and outflow are the ionotropic receptors, which are ligand-gated ion channels that bind specific neurotransmitters, released from the synaptic vesicles of the presynaptic terminal, to trigger the opening of the channels, which serve as conduits for cations that, in turn, initiate action potential across the post synaptic terminals of normally functioning neurons.[7]
The key player in the dramatic process of cationic influx is glutamate, an excitatory neurotransmitter that triggers excitotoxicity during anoxic depolarization.[8] A number of ionotropic receptors have been identified as contributing to anoxic depolarization of nerve cell membranes. They include the NMDA receptors, AMPA receptors, P2X7 purinergic receptors, pannexin channels (Panx1), transient receptor potential (TRP) channels, and acid-sensing ion channels (ASICs).[1]
During brain ischemia, glutamate is released in excess from the presynaptic terminal, leading to the uncontrollable opening of the glutamate receptors, including the NMDA and AMPA receptors, which allows for an excessive influx of Ca2+ into the intracellular environment. Purinergic and NMDA receptors activate the pannexin-1 channels, which become hyperactive and allow the release of ATP from the intracellular environment. As the extracellular glutamate and ATP increase, several complexes are activated and converge into apoptotic and necrotic cascade pathways, which cause neuronal damage and death.[1]
Post-anoxic depolarization: downstream neuronal damage
.jpg.webp)
In the aftermath of anoxic depolarization, at the region of infarction, the release of glutamate and aspartate into the extracellular space causes an uncontrollable intracellular mobilization of Ca2+, mainly through the NMDA receptors.[9] This is a critical stage in the development of neuronal damage, because it is the Ca2+ overload that gives rise to several downstream cascades of events that lead to necrotic neuronal death, or to apoptosis, including free radical and nitric oxide productions that cause damage to the membrane.[10]
Another cytotoxic event that follows anoxic depolarization is lactate accumulation and acidosis as a result of glycolysis, which causes damage to the mitochondria.[10] Ischemic insult also causes blood-brain barrier disruption.[9] Other consequential damage that occurs includes lipolysis, proteolysis, cell swelling, microtubule disaggregation, and DNA fragmentation.[5]
Selective vulnerability
Neurons are more susceptible to brain ischemia than the supporting glial cells, because neurons have higher energy demand, conduct an action potential, and produce glutamate, whereas glial cells lack those properties. Yet neurons differ among themselves in their sensitivity to ischemia, depending on the specific properties they exhibit, relating to their locations in the brain.[11]
Selective vulnerability is how some parts of the brain are more sensitive to anoxia than others, and thus to ischemic insult.[10] Anoxia-prone cells in the brain include the hippocampal pyramidal cells of CA1, cerebellar purkinje cells, pyramidal neocortical neurons in some layers, basal ganglia, reticular neurons of the thalamus, and brainstem neurons.[12]
While basal ganglia, cerebellar purkinje cells, hippocampal, and neocortical cells are more vulnerable to transient ischemic attack (TIA), brainstem and thalamic reticular neurons are more vulnerable to prolonged ischemic attack (stroke proper).[11] Meanwhile, the hippocampal pyramidal cells have been identified as the most vulnerable cells to ischemia.[12] One possible explanation for why selective vulnerability exists attributes the phenomenon to the different amounts of glutamate produced by different neurons, since it is glutamate release to the synaptic cleft that triggers Ca2+ influx, which in turn triggers biochemical processes that damage the neurons.[11] In other research, variation in the expression of immediate early gene and heat shock protein was identified as causing selective vulnerability.[12]
Anoxic-tolerance mechanisms
Metabolic depression
The painted turtle (Chrysemys picta) uses the mechanism of metabolic depression to combat oxygen depletion.[13] Within a few minutes of anoxia onset in the turtle's brain there is decreased cerebral blood flow that eventually ceases. Meanwhile, glycolysis is stimulated to maintain a near optimum ATP production.[3] This compensatory stimulation of glycolysis occurs because, in the turtle's brain, cytochrome a and a3 have a low affinity for oxygen.[13] Anaerobic glycolysis leads to lactate overload, which the turtle buffers to some extent by increased shell and bone CaCO3 production.[3]
However, glycolysis is not efficient for ATP production, and in order to maintain an optimum ATP concentration, the turtle's brain reduces its ATP consumption by suppressing its neuronal activity and gradually releasing adenosine. This re-establishes the ATP consumption/production balance, which is then maintained by reducing ion conductance and releasing GABA. The decrease in neuronal activity renders the turtle comatose for the duration of anoxia.[14]
Pasteur effect
Another anoxia-tolerant animal that is commonly used as a model to study anoxia in the mammalian brain is the crucian carp, which can survive at even more extreme anoxic conditions than the painted turtle can. Unlike C. picta, which takes such drastic measures in becoming comatose to maintain an optimum ATP concentration, the crucian carp does not become comatose in anoxia. Instead, it stays active by maintaining its normal cardial output as well as increasing its cerebral blood flow.[5] Even though glycolysis is stimulated early in anoxia in both the crucian carp and C. picta, the crucian carp is able to stay active because of its capability to re-route the glycolytic pathway such that lactate is converted into ethanol, which can then be released into the water via the gills, thus preventing lactate overload and acidosis.[3]
Since the crucian carp has a more efficient strategy to prevent lactate buildup than C. picta, the initial glycolysis continues without ceasing, a process called the Pasteur effect.[14] In order to keep up with this fast glucose metabolism via glycolysis, as well as maintain the balance between ATP production and consumption, the crucian carp moderately suppresses its motor activities, releases GABA, and selectively suppresses some unnecessary sensory functions.[14] Crucian carp also counteracts the damaging effects of anoxia by swimming into cooler water, a phenomenon known as voluntary hypothermia.[3]
Tolerance in mammalian neonates
The brains of several mammalian neonates have been identified as able to confer resistance to anoxia in a fashion similar to that of the anoxic-tolerant aquatic organisms.[13] This is still a relatively new area of study that could have a clinical significance in combating stroke in humans. A study that looked into anoxic-tolerance in newborn mammals identified two main ways in which they cope with acute hypoxia. While most newborns, preferentially, depress their metabolic rate to conserve energy during anoxia, some mammalian newborns—such as the pig, the deer, and other animals in their class, which are capable of a high degree of independent activity from birth—employ hyperpnoea (abnormally rapid or deep breathing).[15] Why metabolic depression is less effective in adult mammals, compared to neonates, is unclear at the moment. Due to ethical issues, anoxic-tolerance has not been tested in human neonates.
Research: neuroprotective agents
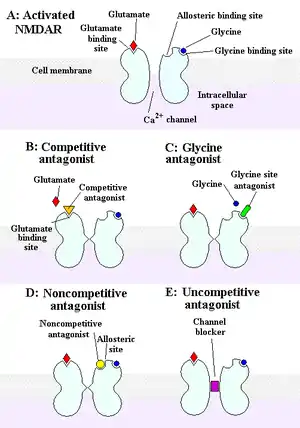
Currently, there is no effective way to combat stroke. The only FDA-approved drug to treat stroke is a clot-dissolving, genetically-engineered enzyme called tissue plasminogen activator, which must be administered within 9 hours of the onset of symptoms, in order to be effective in reducing damage following ischemic stroke.[16]
Many clinical trials have failed in an attempt to develop effective neuroprotective drugs to combat stroke, perhaps because those drugs only deal with one aspect of stroke, and therefore neglect the fact that stroke is a multifaceted problem. Some of the potential treatments for stroke that have been tested by a number of researchers, using several animal models, involve sigma-1 receptor ligands, to modulate Ca2+ release, NMDA receptor antagonists, to prevent Ca2+ overload, and ion channel blockers, to prevent excessive ion fluxes.
See also
References
- Weilinger NL, Maslieieva V, Bialecki J, Sridharan SS, Tang PL, Thompson RJ (2013). "Ionotropic receptors and ion channels in ischemic neuronal death and dysfunction". Acta Pharmacol Sin. 34 (1): 39–48. doi:10.1038/aps.2012.95. PMC 4086487. PMID 22864302.
- Stys, P. (1998). "Anoxic and ischemic injury of myelinated axons in CNS white matter: from Mechanistic Concepts to Therapeutics". Journal of Cerebral Blood Flow and Metabolism. 18 (1): 2–25. doi:10.1097/00004647-199801000-00002. PMID 9428302.
- Nilsson, G.; Lutz, P (2004). "Anoxia tolerant brains". Journal of Cerebral Blood Flow and Metabolism. 24 (5): 475–486. doi:10.1097/00004647-200405000-00001. PMID 15129179.
- Purves, Dale; Augustine, G. J.; Fitzpatrick, D.; Hall, W. C.; LaMantia, A.; McNamara, J. O.; White, L. E. (2008). "Neural Signaling". Neuroscience (4th ed.). Sunderland, MA: Sinauer. pp. 23–207.
- Lutz, P. L.; Nilsson, G. E. (1997). Neuroscience intelligence unit: The Brain Without Oxygen (2nd ed.). Austin, TX: Landes Bioscience and Chapman & Hall. pp. 1–207.
- Kochlamazashvili, G; Haucke, V (2013). "A dual role of SNAP-25 as carrier and guardian of synaptic transmission". EMBO Reports. 14 (7): 579–580. doi:10.1038/embor.2013.74. PMC 3701241. PMID 23732543.
- Goyal, R; Chaudhury, A (2013). "Structure activity relationship of synaptic and junctional neurotransmission". Autonomic Neuroscience: Basic and Clinical. 176 (1–2): 11–31. doi:10.1016/j.autneu.2013.02.012. PMC 3677731. PMID 23535140.
- Madry, C; Haglerød, C; Attwell, D (2010). "The role of pannexin hemichannels in the anoxic depolarization of hippocampal pyramidal cells". Brain. 133 (Pt 12): 3755–3763. doi:10.1093/brain/awq284. PMC 2995884. PMID 20940167.
- Zhao, H; Steinberg, G.; Sapolsky, R (2007). "General versus specific actions of mild-moderate hypothermia in attenuating cerebral ischemic damage". Journal of Cerebral Blood Flow and Metabolism. 27 (12): 1879–1894. doi:10.1038/sj.jcbfm.9600540. PMID 17684517.
- Huang, B; Castillo, M (2008). "Hypoxic-ischemic brain injury: imaging findings from birth to adulthood". Radiographics. 28 (2): 417–439. doi:10.1148/rg.282075066. PMID 18349449.
- Agamanolis, D. "Chapter 2: Cerebral Ischemia and Stroke". Neuropathology. Retrieved 4 November 2013.
- Busl, K; Greer, D (2010). "Hypoxic-ischemic brain injury: pathophysiology, neuropathology and mechanisms". NeuroRehabilitation. 26 (1): 5–13. doi:10.3233/NRE-2010-0531. PMID 20130351.
- Lutz, P. L (1992). "Mechanisms for Anoxic Survival in the Vertebrate Brain". Annual Review of Physiology. 54: 601–618. doi:10.1146/annurev.ph.54.030192.003125. PMID 1348613.
- John W. Thompson; Göran E. Nilsson; Miguel A. Perez-Pinzon (2013). "2: Anoxia Resistance in Lower and Higher Vertebrates". In Jeffrey M. Gidday; Miguel A. Perez-Pinzon; John H. Zhang (eds.). Innate Tolerance in the CNS: Translational Neuroprotection by Pre- and Post-Conditioning. New York: Springer New York. pp. 19–35. ISBN 978-1-4419-9694-7.
- Mortola, J (1999). "How newborn mammals cope with hypoxia". Respiration Physiology. 116 (2–3): 95–103. doi:10.1016/S0034-5687(99)00038-9. PMID 10487295.
- Behensky, A; Cortes-Salva, M.; Seminerio, M.; Matsumoto, R.; Antilla, J.; Cuevas, J. (2013). "In vitro evaluation of guanidine analogs as sigma receptor ligands for potential anti-stroke therapeutics". The Journal of Pharmacology and Experimental Therapeutics. 344 (1): 155–166. doi:10.1124/jpet.112.199513. PMC 3533416. PMID 23065135.
External links
- The Ischemic Penumbra
- Lai TW, Shyu WC, Wang YT (2011). "Stroke intervention pathways: NMDA receptors and beyond". Trends Mol Med. 17 (5): 266–75. doi:10.1016/j.molmed.2010.12.008. PMID 21310659.