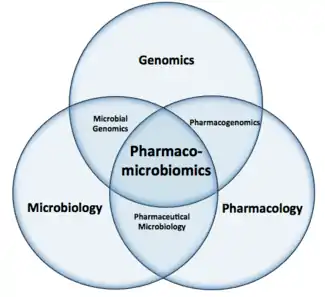
Pharmacomicrobiomics
Pharmacomicrobiomics, first used in 2010, is defined as the effect of microbiome variations on drug disposition, action, and toxicity.[1] Pharmacomicrobiomics is concerned with the interaction between xenobiotics, or foreign compounds, and the gut microbiome. It is estimated that over 100 trillion prokaryotes representing more than 1000 species reside in the gut.[2][3] Within the gut, microbes help modulate developmental, immunological and nutrition host functions.[4] The aggregate genome of microbes extends the metabolic capabilities of humans, allowing them to capture nutrients from diverse sources.[5] Namely, through the secretion of enzymes that assist in the metabolism of chemicals foreign to the body, modification of liver and intestinal enzymes, and modulation of the expression of human metabolic genes, microbes can significantly impact the ingestion of xenobiotics.[6]
Efforts to understand the interaction between specific xenobiotics and the microbiome have traditionally involved the use of in vivo as well as in vitro models.[7] Recently, next generation sequencing of genomic DNA obtained from a community of microbes has been used to identify organisms within microbial communities, allowing for accurate profiles of the composition of microbes within an environment. Initiatives such as the Human Microbiome Project (HMP) have aimed to characterize the microbial composition of the oral, gut, vaginal, skin and nasal environments.[8] This and other microbiome characterization projects have accelerated the study of pharmacomicrobiomics. An extensive understanding of the microbiome in the human body can lead to the development of novel therapeutics and personalized drug treatments that are not potentiated or activated by processes carried out by the microbiome.
History
In a 1973 paper, Ronald Scheline stated that the gastrointestinal microbiome has the ability to act as an organ with metabolic potential at least equal to the liver.[9] Since then, the importance of the human microbiome in mediating health and disease has been acknowledged, and specific interactions between xenobiotics and microbes have been characterized using in vitro or in vivo methods. However, few studies have taken into account the complete metabolic profile, leading some to say that the microbiome's cumulative role in xenobiotic metabolism and toxicology has largely remained unexplored.[10] It is reported that 84% of the top-selling pharmaceuticals in the US and Europe are administered orally, making it the most common mode of drug administration.[11] The implication of this is that a large proportion of drugs, especially those that are lowly soluble and permeable ones, encounter the microbiome and are subject to reductive and hydrolytic reactions.[12]
Sequencing technologies such as 16S rRNA shotgun metagenomic sequencing have facilitated the rapid expansion of the pharmacomicrobiomics field by capturing organismal diversity in microbial communities. The Human Microbiome Project and METAgenomics of the Human Intestinal Tract (MetaHIT), established in 2007 and 2008, respectively, aimed to characterize the variation in human microbiomes.[13] These large scale projects are foundational to pharmacomicrobiomic studies, as they allow for the generation of statistic models that can take into account variation in microbial composition across individuals.
Methods to elucidate microbiome composition
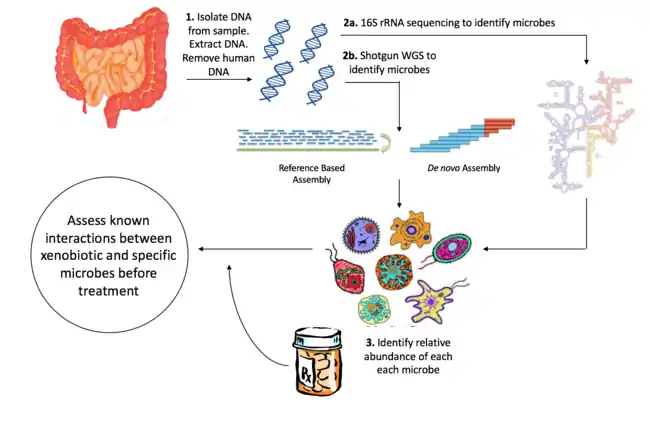
Animal models
Interactions between xenobiotics and the host microbiome have primarily been assessed through the use of in vivo animal models, as it is difficult to model the natural human gut. In general, the pattern of bacterial colonization is the same in different animals, with both pH and the number of microorganisms gradually increasing from the small intestine towards the ileo-caecal junction of the large intestine.[15] Germ-free rats colonized with human faecal matter are generally regarded as the gold standard in animal modeling of gut microbial environment.[16] However, enzyme activity can vary greatly between organisms.
In vitro models
Microbes found in human fecal samples are fairly representative of the gut microbiome, and are used frequently in in vitro cultures. A variety of in vitro microbial modelling techniques have also been developed. Static batch culturing consists of plating bacteria without replenishing the media at regular intervals.[17] Semi-continuous culture systems allow for the addition of medium without disrupting bacterial growth, and include pH control capabilities.[18] The continuous culture system more closely resembles that of the gut, as it continuously replenishes and removes culture medium.[19] The simulator of the human intestinal microbial system (SHIME) models the small and large intestine through the use of a five-stage reactor, and includes numerous ports for continuous monitoring of pH and volume.[20] Most recently, researchers improved on SHIME by including a computer controlled peristaltic wave to circulate chyme throughout the apparatus.[21] These technologies have given researchers close control over the culturing environment, facilitating the discovery of interactions between xenobiotics and microbes.
16S rRNA Sequencing
16S ribosomal RNA is the most common housekeeping genetic marker for classifying and identifying bacterial species, as it is present in all bacterial species, has an identical function in most organisms, and is large enough (~1,500 bp) to capture sufficient variation to distinguish bacteria.[22] The sequence of 16S rRNA consists of highly conserved sequences which alternate with nine windows of "hypervariable regions".[23] This allows universal primers to be used to sequence many species at a time, and provides the possibility of distinguishing bacteria given the variable regions alone. Many papers suggest that 16S rRNA gene sequencing provides genus identification in >90% of cases, but species level identification in approximately ~65 to 83% of cases.[24] The Ribosomal Database Project (RDP)[25] and SILVA databases contain sequence information for rRNA in bacteria, eukarya and archaea.[26]
Shotgun sequencing
Advances in high-throughput sequencing has facilitated shotgun metagenome sequencing (SMS), a technology that provides a broader characterization of microbial samples by sequencing a larger number of genes in each organism. SMS involves collecting microbial samples from the environment, isolating DNA, shearing the DNA into small fragments, and then performing whole genome sequencing (WGS). Reads can be assembled de novo or using reference genomes.[27] However, SMS is not without limitations. Reads may overlap and prevent accurate alignment to reference genomes. In addition, reads may be contaminated by human DNA sequence, confounding results. In reference-based assembly, reads may also be biased towards species which have publicly available reference genomes.
Composition of the microbiome
Gut
Within the intestines, the majority of microbes can be found in the large intestine, where the pH is higher and more conducive to survival. These bacteria are often more efficient than our own digestive enzymes, and function to digest protein and carbohydrates.[28] The results of over 690 human microbiomes have shown that the majority of bacteria of the gut microbiome belongs to four phyla: Firmicutes, Bacteroidetes, Actinobacteria, and Proteobacteria.[29]
Vagina
The vagina possesses over 200 phylotypes, the most predominant belonging to the phyla Firmicutes, Bacteroidetes, Actinobacteria, and Fusobacteria.[30] The secretion of lactic acid and hydrogen peroxide by Lactobacillus sp. can lower the pH, increasing the concentration of bacteria that cause bacterial vaginosis.
Placenta
The first profile of microbes in healthy term pregnancies identified non-pathogenic commensal microbiota from the Firmicutes, Tenericutes, Proteobacteria, Bacteroidetes, and Fusobacteria phyla.[31]
Oral cavity
Through the HMP, nine intraoral sites were in investigated, and found to be enriched in over 300 genera belonging to more than 20 bacterial phyla.[32]
Human Microbiome Project
The Human Microbiome Project (HMP) was established in 2008 by the US National Institutes of Health (NIH). The overarching goal is to establish a comprehensive characterization of the human microbiota and its role in human health and disease, as well as to develop datasets and tools that scientists can use to study microbial populations.[33] The specific initiatives are as follows:
- Develop a reference set of microbial genome sequences for an initial characterization of the human microbiome.
- Elucidate the relationship between disease and changes in the human microbiome.
- Develop technologies for computational analysis, namely methods for sequencing individual microbes or all members of complex populations simultaneously.
- Establish a Data Analysis and Coordinating Center to provide publicly available information about the project, outcomes, and raw data.
- Establish research repositories to store materials and reagents used in the HMP. This includes cultured organisms and metagenomic DNA samples.
- Examine ethical, legal, and social implications of HMP research.
The primary means of characterization is through 16S rRNA sequencing and shotgun metagenomic sequencing. Body sites that are sampled include skin, oral cavity, gut, vagina and nasal cavity.[34] The HMP website includes sequence, metabolic reconstruction, and community profile data. These datasets have been used to associate certain clinical variables with microbiome composition[35][36]
Known Drug Interactions
Microbiota-mediated interference in xenobiotic activity
The microbiome can significantly affect the potency of a pharmaceutical drug. Even though most drugs are absorbed in the upper part of the large intestine, long-acting drugs that are exposed to the microbe-rich area of the lower intestine can be affected by microbial metabolism. For instance, chloramphenicol may cause bone marrow aplasia following oral administration, due to the presence of coliforms that convert chloramphenical to its toxic form, known as p-aminophenyl-2-amin-1,2-propanediol.[37] In addition, altered abundances of Eggerthella lenta between populations have been found to affect the metabolism of digoxin, potentiating both its activity and toxicity.[38] A non-exhaustive list of drugs and the microbiota’s role in potentiating/increasing their effect is provided below.
| Drug | Pharmacological effect | Effect of microbiota on clinical outcome | Reference |
|---|---|---|---|
| Acetaminophen | Analgesic and antipyretic | Increased clinical effect and toxicity | [39] |
| Chloramphenicol | Antibiotic | Increase toxicity | [40] |
| Digoxin | Cardiotonic | Decrease toxicity and activity | [41] |
| Flucytosine | Antifungal | Decrease effect | [42] |
| Metronidazole | Antibiotic | Provide resistance to the antimicrobial/antifungal effect. Also lowers the effect by stimulating metabolism. | [43] |
| Sulfinpyrazone | Antibiotic | Activate the drug | [44] |
| Sulindac | Nonsteroidal anti-inflammatory drug | Activate the drug | [45] |
Xenobiotic mediated interference in microbiome composition
Even though pharmacomicrobiomics is often interpreted as the impact the microbiome has on xenobiotic metabolism, the term can also encompass the effects of xenobiotics on the microbiome and microbial genes. The impact of antibiotics on the human microbiome has been well studied. It has been shown that antibiotic therapies not only target a specific pathogen, but also the commensal inhabitants of a host.[46] Evidence suggests that commensal bacteria levels in some cases are not normalized after antibiotic treatment, and in fact may be negatively affected for extended periods of time.[47] A study which assessed the oral and gut microbes before, immediately after, and up to 12 months after exposure to antibiotics, found that the microbiome can be altered for over 12 months.[48] Since the microbiome composition can be altered by antibiotics, this implies positive selection for resistant opportunistic pathogens, which can cause acute disease.[49]
The PharmacoMicrobiomics Web Portal
The PharmacoMicrobiomics Web Portal[50] is a student-led initiative to explore how microbes modulate drugs[51] that is intended for bioinformaticians, microbial geneticists, and drug developers. The goal of the project is to mine literature data and extract microbe-drug interactions, including information about drug classes, microbial families, and body systems. Furthermore, the portal includes a relational database with information on microbial composition at different body sites and their specific effects on drug pharmacokinetics and pharmacodynamic properties.
Personalized Medicine
Personalized medicine in the context of pharmacomicrobiomics refers to the ability to predict an individual’s response to a xenobiotic based on the composition of their gut microbiome. However, current omics approaches investigating microbiome composition using metagenomic sequencing after xenobiotic treatment are sparse. Instead, research efforts have focused predominantly on modeling changes in microbial composition in different disease states.[52] Future research efforts should combine knowledge relating to what microbes preferentially metabolize certain compounds (garnered from in vitro studies) with the identification of species abundance to predict drug tolerance in patients. However, modeling a microbe’s interaction with a particular xenobiotic may not stably predict interactions, as the genomes of microbes are continually reshuffled through horizontal gene transfer. Considering this, approaches that target individual gene/transcript/protein signatures rather than individual microbes will likely lead to more widely applicable personalized approaches.[53]
Limitations
The limitations of pharmacomicrobiomics primarily arise from the uncertainty associated with metagenomic profiling. Namely, short reads obtained by shotgun sequencing can be difficult to align to reference genomes since many organism have homologous sequences. In addition, 16S rRNA sequencing cannot consistently resolve species identity, a finding that casts doubt on species identities in metagenomic samples. Limitations also arise from differing study designs, as unique approaches to identifying the nature of the xenobiotic-microbiome interactions are often taken. For instance, because pharmacomicrobiomics very broadly denotes the association between xenobiotics and the microbiome, the extent to which studies profile the genetics of the microbiome can vary significantly. Studies aiming to characterize organism identity, but not gene identity or copy number may elect to use 16S shotgun sequencing as opposed to SMS. Conversely, studies aiming to identify genes and their products rather than organism identity may elect WMGS coupled with transcriptomic analysis. Initially, these differences may mean that researchers wanting to investigate publicly available data may have to target their research questions to fit the data at hand.
References
- Rizkallah, M. R.; Saad, R.; Aziz, R. K. (2010). "The Human Microbiome Project, Personalized Medicine and the Birth of Pharmacomicrobiomics". Current Pharmacogenomics and Personalized Medicine. 8 (3): 12. doi:10.2174/187569210792246326.
- Ley, R; Turnbaugh, P; Klein, S; Gordon, J (2006). "Microbial ecology: human gut microbes associated with obesity". Nature. 444 (7122): 1022–1023. doi:10.1038/nature4441021a.
- Arumugam, M; Raes, J; Pelletier, E; et al. (2013). "Enterotypes of the human gut microbiome". Nature. 473 (7346): 174–180. doi:10.1038/nature09944.Enterotypes.
- Egert, M; De Graaf, AA; Smidt, H; De Vos, WM; Venema (2006). "Functional microbiomics of the human colon". Trends Microbiol. 14 (2): 86–91. doi:10.1016/j.tim.2005.12.007.
- Haiser, HJ; Turnbaugh, PJ (2013). "Developing a metagenomic view of xenobiotic metabolism". Pharmacol. Res. 69 (1): 21–31. doi:10.1016/j.phrs.2012.07.009. PMC 3526672.
- Saad, R; Rizkallah, MR; Aziz, RK (2012). "Gut Pharmacomicrobiomics: the tip of an iceberg of complex interactions between drugs and gut-associated microbes". Gut Pathog. 4 (1): 16. doi:10.1186/1757-4749-4-16.
- Sousa, T; Paterson, R; Moore, V; Carlsson, A; Abrahamsson, B; Basit, AW (2008). "The gastrointestinal microbiota as a site for the biotransformation of drugs". Int J Pharm. 363 (1–2): 1–25. doi:10.1016/j.ijpharm.2008.07.009.
- Notes, S (2012). "A framework for human microbiome research". Nature. 486 (7402): 215–221. doi:10.1038/nature11209.
- Scheline, RR (1973). "Metabolism of foreign compounds by gastrointestinal microorganisms". Pharmacol Rev. 25 (4): 451–523.
- Wilson, ID; Nicholson, JK (2016). "Gut microbiome interactions with drug metabolism, efficacy, and toxicity". Transl. Res. 179: 204–222. doi:10.1016/j.trsl.2016.08.002. PMC 5718288.
- Lennernäs, H; Abrahamsson, B (2005). "The use of biopharmaceutic classification of drugs in drug discovery and development: current status and future extension". J Pharm Pharmacol. 57 (3): 273–285. doi:10.1211/0022357055263.
- Sousa, T; Paterson, R; Moore, V; Carlsson, A; Abrahamsson, B; Basit, AW (2008). "The gastrointestinal microbiota as a site for the biotransformation of drugs". Int J Pharm. 363 (1–2): 1–25. doi:10.1016/j.ijpharm.2008.07.009.
- https://commonfund.nih.gov/hmp/overview, http://www.gutmicrobiotaforhealth.com/en/metahit/
- http://rna.ucsc.edu/rnacenter/images/figs/thermus_16s_2ndry.jpg
- Sousa, T; Paterson, R; Moore, V; Carlsson, A; Abrahamsson, B; Basit, AW (2008). "The gastrointestinal microbiota as a site for the biotransformation of drugs". Int J Pharm. 363 (1–2): 1–25. doi:10.1016/j.ijpharm.2008.07.009.
- Sousa, T; Paterson, R; Moore, V; Carlsson, A; Abrahamsson, B; Basit, AW (2008). "The gastrointestinal microbiota as a site for the biotransformation of drugs". Int J Pharm. 363 (1–2): 1–25. doi:10.1016/j.ijpharm.2008.07.009.
- Rowland, I. (2012-12-02). Role of the Gut Flora in Toxicity and Cancer. ISBN 9780323147057.
- Rumney, CJ; Rowland, IR (1992). "In vivo and in vitro models of the human colonic flora". Crit Rev Food Sci Nutr. 31 (4): 299–331. doi:10.1080/10408399209527575. PMID 1581008.
- Marsh, PD (1995). "The role of continuous culture in modelling the human microflora". J Chem Technol Biotechnol. 64 (1): 1–9. doi:10.1002/jctb.280640102.
- Molly K, Woestyne M Vande, Smet I De, Verstraete W. Microbial Ecology in Health and Disease Validation of the Simulator of the Human Intestinal Microbial Ecosystem ( SHIME ) Reactor Using Microorganism-associated Activities Validation of the Simulator of the Human Intestinal Microbial Ecosystem ( SHIME ) R. 2009;2235(March 2017).
- Minekus M, Marteau P, Havenaar R, Huis in ’t Veld JHJ. A multicompartmental dynamic computer-controlled model simulating the stomach and small intestine. Altern to Lab Anim. 1995;23(August 2015):197-209.
- Janda, JM; Abbott, SL (2007). "16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: Pluses, perils, and pitfalls". J Clin Microbiol. 45 (9): 2761–2764. doi:10.1128/JCM.01228-07.
- Chakravorty, S; Helb, D; Burday, M; Connell, N (2007). "A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria". J Microbiol Methods. 69 (2): 330–339. doi:10.1016/j.mimet.2007.02.005.A.
- Drancourt, M; Bollet, C; Carlioz, A; Martelin, R; Gayral, JP; Raoult, D (2000). "16S ribosomal DNA sequence analysis of a large collection of environmental and clinical unidentifiable bacterial isolates". J Clin Microbiol. 38 (10): 3623–3630. doi:10.1073/pnas.0504930102.
- Cole, JR; Wang, Q; Fish, JA; et al. (2014). ""Ribosomal Database Project " Data and tools for high throughput rRNA analysis". Nucleic Acids Res. 42 (D1): D1. doi:10.1093/nar/gkt1244.
- Quast, C; Pruesse, E; Yilmaz, P; et al. (2013). ""The SILVA ribosomal RNA gene database project " Improved data processing and web-based tools". Nucleic Acids Res. 41 (D1): D1. doi:10.1093/nar/gks1219.
- Sharpton, TJ (2014). "An introduction to the analysis of shotgun metagenomic data". Front Plant Sci. 5: 209. doi:10.3389/fpls.2014.00209.
- Scheline, RR (1973). "Metabolism of foreign compounds by gastrointestinal microorganisms". Pharmacol Rev. 25 (4): 451–523.
- Notes, S (2012). "A framework for human microbiome research". Nature. 486 (7402): 215–221. doi:10.1038/nature11209.
- Ravel, J; Gajer, P; Abdo, Z; et al. (2011). "Vaginal microbiome of reproductive-age women". Proc Natl Acad Sci. 108: 4680–4687. doi:10.1073/pnas.1002611107.
- Aagaard, K; Ma, J; Antony, KM; Ganu, R; Petrosino, J; Versalovic, J (2014). "The placenta harbors a unique microbiome". Sci Transl Med. 6 (237): 237–65. doi:10.1126/scitranslmed.3008599. PMC 4929217.
- Zhou, Y; Gao, H; Mihindukulasuriya, KA; et al. (2013). "Biogeography of the ecosystems of the healthy human body". Genome Biol. 14 (1): R1. doi:10.1186/gb-2013-14-1-r1.
- Notes, S (2012). "A framework for human microbiome research". Nature. 486 (7402): 215–221. doi:10.1038/nature11209.
- Notes, S (2012). "A framework for human microbiome research". Nature. 486 (7402): 215–221. doi:10.1038/nature11209.
- Rogers GB, Narkewicz MR, Hoffman LR. "The CF gastrointestinal microbiome: Structure and clinical impact. Pediatr Pulmonol. 2016;51(May):S35-S44. doi:10.1002/ppul.23544.
- Lloyd-Price, J; Abu-Ali, G; Huttenhower, C (2016). "The healthy human microbiome". Genome Med. 8: 51. doi:10.1186/s13073-016-0307-y.
- Grundmann, O; Yoon, SL (2010). "Irritable bowel syndrome: Epidemiology, diagnosis and treatment: An update for health-care practitioners". J Gastroenterol Hepatol. 25 (4): 691–699. doi:10.1111/j.1440-1746.2009.06120.x.
- Mathan, VI; Wiederman, J; Dobkin, JF; Lindenbaum, J (1989). "Geographic differences in digoxin inactivation, a metabolic activity of the human anaerobic gut flora". Gut. 30 (7): 971–977. doi:10.1136/gut.30.7.971. PMC 1434295.
- Clayton, TA; Baker, D; Lindon, JC; Everett, JR; Nicholson, JK (2009). "Pharmacometabonomic identification of a significant host-microbiome metabolic interaction affecting human drug metabolism". Proc Natl Acad Sci U S A. 106 (34): 14728–14733. doi:10.1073/pnas.0904489106.
- Grundmann, O; Yoon, SL (2010). "Irritable bowel syndrome: Epidemiology, diagnosis and treatment: An update for health-care practitioners". J Gastroenterol Hepatol. 25 (4): 691–699. doi:10.1111/j.1440-1746.2009.06120.x.
- Mathan, VI; Wiederman, J; Dobkin, JF; Lindenbaum, J (1989). "Geographic differences in digoxin inactivation, a metabolic activity of the human anaerobic gut flora". Gut. 30 (7): 971–977. doi:10.1136/gut.30.7.971. PMC 1434295.
- Vermes, A; Kuijper, EJ; Guchelaar, HJ; Dankert, J (2003). "An in vitro study on the active conversion of flucytosine to fluorouracil by microorganisms in the human intestinal microflora". Chemotherapy. 49 (1–2): 17–23. doi:10.1159/000069784.
- Steffens, LS; Nicholson, S; Paul, L V.; Nord, CE; Patrick, S; Abratt, VR. (2010). "Bacteroides fragilis RecA protein overexpression causes resistance to metronidazole". Res Microbiol. 161 (5): 346–354. doi:10.1016/j.resmic.2010.04.003.
- Strong, HA; Renwick, AG; George, CF; Liu, YF; Hill, MJ (1987). "The reduction of sulphinpyrazone and sulindac by intestinal bacteria". Xenobiotica. 17 (6): 685–696. doi:10.3109/00498258709043976.
- Strong, HA; Renwick, AG; George, CF; Liu, YF; Hill, MJ (1987). "The reduction of sulphinpyrazone and sulindac by intestinal bacteria". Xenobiotica. 17 (6): 685–696. doi:10.3109/00498258709043976.
- Jernberg, C; Löfmark, S; Edlund, C; Jansson, JK (2010). "Long-term impacts of antibiotic exposure on the human intestinal microbiota". Microbiology. 156 (11): 3216–3223. doi:10.1099/mic.0.040618-0.
- Jernberg, C; Löfmark, S; Edlund, C; Jansson, JK (2010). "Long-term impacts of antibiotic exposure on the human intestinal microbiota". Microbiology. 156 (11): 3216–3223. doi:10.1099/mic.0.040618-0.
- Zaura, E; Brandt, BW; Joost, M; et al. (2015). ""Same Exposure but Two Radically Different Responses to Antibiotics " Resilience of the Salivary Microbiome versus Long-Term Microbial". Am Soc Microbiol. 6 (6): 1–11. doi:10.1128/mBio.01693-15.Editor.
- Francino, MP (2016). "Antibiotics and the human gut microbiome: Dysbioses and accumulation of resistances". Front Microbiol. 6. doi:10.3389/fmicb.2015.01543.
- R. Rizkallah, Mariam; Gamal-Eldin, Soha; Saad, Rama; Aziz, Ramy K. (2012). "The PharmacoMicrobiomics Portal: A Database for Drug-Microbiome Interactions". Current Pharmacogenomics and Personalized Medicine. 10 (3): 195–203. doi:10.2174/187569212802510030.
- Aziz, RK; Saad, R; Rizkallah, MR (2011). "PharmacoMicrobiomics or how bugs modulate drugs: an educational initiative to explore the effects of human microbiome on drugs". BMC Bioinformatics. 12 (Suppl 7): A10. doi:10.1186/1471-2105-12-S7-A10.
- Kostic, AD; Xavier, RJ; Gevers, D (2014). "The microbiome in inflammatory bowel disease: Current status and the future ahead". Gastroenterology. 146 (6): 1489–1499. doi:10.1053/j.gastro.2014.02.009. PMC 4034132.
- Kong, SW; Collins, CD; Shimizu-motohashi, Y; et al. (2012). "Characteristics and Predictive Value of Blood Transcriptome Signature in Males with Autism Spectrum Disorders". PLOS ONE. 7: e49475. doi:10.1371/journal.pone.0049475.

{kind=link}