Phi value analysis
Phi value analysis, analysis, or -value analysis is an experimental protein engineering technique for studying the structure of the folding transition state of small protein domains that fold in a two-state manner. The structure of the folding transition state is hard to find using methods like protein NMR or X-ray crystallography because folding transitions states are mobile and partly unstructured by definition. In -value analysis, the folding kinetics and conformational folding stability of the wild-type protein are compared with those of point mutants to find phi values. These measure the mutant residue's energetic contribution to the folding transition state, which reveals the degree of native structure around the mutated residue in the transition state, by accounting for the relative free energies of the unfolded state, the folded state, and the transition state for the wild-type and mutant proteins.

The protein's residues are mutated one by one to identify residue clusters that are well-ordered in the folded transition state. These residues' interactions can be checked by double-mutant-cycle analysis, in which the single-site mutants' effects are compared to the double mutants'. Most mutations are conservative and replace the original residue with a smaller one (cavity-creating mutations) like alanine, though tyrosine-to-phenylalanine, isoleucine-to-valine and threonine-to-serine mutants can be used too. Chymotrypsin inhibitor, SH3 domains, individual domains of proteins L and G, ubiquitin, and barnase have all been studied by analysis.
Mathematical approach
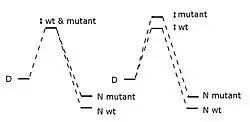
Phi is defined thus:[1]

is the difference in energy between the wild-type protein's transition and denatured state, is the same energy difference but for the mutant protein, and the bits are the differences in energy between the native and denatured state. The phi value is interpreted as how much the mutation destabilizes the transition state versus the folded state.



Though may have been meant to range from zero to one, negative values can appear.[2] A value of zero suggests the mutation doesn't affect the structure of the folding pathway's rate-limiting transition state, and a value of one suggests the mutation destabilizes the transition state as much as the folded state; values near zero suggest the area around the mutation is relatively unfolded or unstructured in the transition state, and values near one suggest the transition state's local structure near the mutation site is similar to the native state's. Conservative substitutions on the protein's surface often give phi values near one. When is well between zero and one, it is less informative as it doesn't tell us which is the case:
- The transition state itself is partly structured; or
- There are two protein populations of near-equal numbers, one kind which is mostly-unfolded and the other which is mostly-folded.
Key assumptions
- Phi value analysis assumes Hammond's postulate, which states that energy and chemical structure are correlated. Though the relationship between the folding intermediate and native state's structures may correlate that between their energies when the energy landscape has a well-defined, deep global minimum, free energy destabilizations may not give useful structural information when the energy landscape is flatter or has many local minima.
- Phi value analysis assumes the folding pathway isn't significantly altered, though the folding energies may be. As nonconservative mutations may not bear this out, conservative substitutions, though they may give smaller energetic destabilizations which are harder to detect, are preferred.
- Restricting to numbers greater than zero is the same as assuming the mutation increases the stability and lowers the energy of neither the native nor the transition state. It is in the same line assumed that interactions that stabilize a folding transition state are like those of the native structure, though some protein folding studies found that stabilizing non-native interactions in a transition state facilitates folding.[3]
Example: barnase
Alan Fersht pioneered phi value analysis in his study of the small bacterial protein barnase.[4][5] Using molecular dynamics simulations, he found that the transition state between folding and unfolding looks like the native state and is the same no matter the reaction direction. Phi varied with the mutation location as some regions gave values near zero and others near one. The distribution of values throughout the protein's sequence agreed with all of the simulated transition state but one helix which folded semi-independently and made native-like contacts with the rest of the protein only once the transition state had formed fully. Such variation in the folding rate in one protein makes it hard to interpret values as the transition state structure must otherwise be compared to folding-unfolding simulations which are computationally expensive.
Variants
Other 'kinetic perturbation' techniques for studying the folding transition state have appeared recently. Best known is the psi () value[6][7] which is found by engineering two metal-binding amino acid residues like histidine into a protein and then recording the folding kinetics as a function of metal ion concentration,[8] though Fersht thought this approach difficult.[9] A 'cross-linking' variant of the -value was used to study segment association in a folding transition state as covalent crosslinks like disulfide bonds were introduced.[10]

Limitations
The error in equilibrium stability and aqueous (un)folding rate measurements may be large when values of for solutions with denaturants must be extrapolated to aqueous solutions that are nearly pure or the stability difference between the native and mutant protein is 'low', or less than 7 kJ/mol. This may cause to fall beyond the zero-one range.[11] Calculated values depend strongly on how many data point are available and lab conditions.[12]
References
- Daggett V, Fersht AR. (2000). Transition states in protein folding. In Mechanisms of Protein Folding 2nd ed, editor RH Pain. Oxford University Press.
- Rios, MA; Daneshi, M; Plaxco, KW (2005). "Experimental investigation of the frequency and substitution dependence of negative phi-values in two-state proteins". Biochemistry. 44 (36): 12160–7. doi:10.1021/bi0505621. PMID 16142914.
- Zarrine-Asfar, Arash; Wallin, Stefan (July 22, 2008). "Theoretical and Experimental Demonstration of the Importance of Specific Nonnative Interactions in Protein Folding" (PDF). Proceedings of the National Academy of Sciences of the United States of America. Natural Academy of Sciences. 105 (29): 9999–10004. doi:10.1073/pnas.0801874105. PMC 2481363. PMID 18626019.
- Matouschek, A; Kellis, JT; Serrano, L; Fersht, AR (1989). "Mapping the transition state and pathway of protein folding by protein engineering". Nature. 340 (6229): 122–6. doi:10.1038/340122a0. PMID 2739734.
- Fersht, AR; Matouschek, A; Serrano, L (1992). "The folding of an enzyme I. Theory of protein engineering analysis of stability and pathway of protein folding". J Mol Biol. 224 (3): 771–82. doi:10.1016/0022-2836(92)90561-W. PMID 1569556.
- Sosnick, TR; Dothager, RS; Krantz, BA (2004). "Differences in the folding transition state of ubiquitin indicated by and analyses". Proc. Natl. Acad. Sci. USA. 101 (50): 17377–17382. doi:10.1073/pnas.0407683101. PMC 536030. PMID 15576508.
- Krantz, BA; Dothager, RS; Sosnick, TR (March 2004). "Discerning the structure and energy of multiple transition states in protein folding using -analysis". J. Mol. Biol. 337: 463–75. doi:10.1016/j.jmb.2004.01.018. PMID 15003460.
- Krantz, BA; Sosnick, TR (2001). "Engineered metal binding sites map the heterogeneous folding landscape of a coiled coil". Nat. Struct. Biol. 8 (12): 1042–1047. doi:10.1038/nsb723. PMID 11694889.
- Fersht, AR (2004). " value versus analysis". Proc. Natl. Acad. Sci. USA. 101 (50): 17327–17328. doi:10.1073/pnas.0407863101. PMC 536033. PMID 15583125.
- Wedemeyer, WJ; Welker, E; Narayan, M; Scheraga, HA (June 2000). "Disulfide bonds and protein folding". Biochemistry. 39 (23): 7032. doi:10.1021/bi005111p. PMID 10841785.
- Sanchez, IE; Kiefhaber, T (2003). "Origin of unusual phi-values in protein folding: evidence against specific nucleation sites". J. Mol. Biol. 334 (5): 1077–1085. doi:10.1016/j.jmb.2003.10.016. PMID 14643667.
- Miguel; Los Rios, A. De; Muraidhara, B.K.; Wildes, David; Sosnick, Tobin R.; Marqusee, Susan; Wittung-Stafshede, Pernilla; Plaxco, Kevin W.; Ruczinski, Ingo (2006). "On the precision of experimentally determined protein folding rates and phi-values". Protein Science. 15 (3): 553–563. doi:10.1110/ps.051870506. PMC 2249776. PMID 16501226.
