SCO1
Protein SCO1 homolog, mitochondrial, also known as SCO1, cytochrome c oxidase assembly protein, is a protein that in humans is encoded by the SCO1 gene.[5][6] SCO1 localizes predominantly to blood vessels, whereas SCO2 is barely detectable, as well as to tissues with high levels of oxidative phosphorylation. The expression of SCO2 is also much higher than that of SCO1 in muscle tissue, while SCO1 is expressed at higher levels in liver tissue than SCO2. Mutations in both SCO1 and SCO2 are associated with distinct clinical phenotypes as well as tissue-specific cytochrome c oxidase (complex IV) deficiency.[7][8][9]


Structure
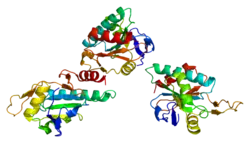
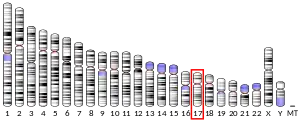
SCO1 is located on the p arm of chromosome 17 in position 13.1 and has 6 exons.[6] The SCO1 gene produces a 33.8 kDa protein composed of 301 amino acids.[10][11] The protein is a member of the SCO1/2 family. It contains 3 copper metal binding sites at positions 169, 173, and 260, a transit peptide, a 25 amino acid topological domain from positions 68-92, a 19 amino acid helical transmembrane domain from positions 93-111, and a 190 amino acid topological domain from positions 112-301 in the mitochondrial intermembrane. Additionally, SCO1 has been predicted to contain 10 beta-strands, 7 helixes, and 2 turns and is a single-pass membrane protein.[8][9]
Function
Mammalian cytochrome c oxidase (COX) catalyzes the transfer of reducing equivalents from cytochrome c to molecular oxygen and pumps protons across the inner mitochondrial membrane. In yeast, 2 related COX assembly genes, SCO1 and SCO2 (synthesis of cytochrome c oxidase), enable subunits 1 and 2 to be incorporated into the holoprotein. This gene is the human homolog to the yeast SCO1 gene.[6] It is predominantly expressed in muscle, heart, and brain tissues, which are also known for their high rates of oxidative phosphorylation.[5] SCO1 is a copper metallochaperone that is located in the inner mitochondrial membrane and is important for the maturation and stabilization of cytochrome c oxidase subunit II (MT-CO2/COX2). It plays a role in the regulation of copper homeostasis by controlling the localization and abundance of CTR1 and is responsible for the transportation of copper to the Cu(A) site on MT-CO2/COX2.[12][8][9][13]
Clinical relevance
Mutations in the SCO1 gene are associated with hepatic failure and encephalopathy resulting from mitochondrial complex IV deficiency also known as cytochrome c oxidase deficiency. This is a disorder of the mitochondrial respiratory chain with heterogeneous clinical manifestations, ranging from isolated myopathy to severe multisystem disease affecting several tissues and organs. Features include hypertrophic cardiomyopathy, hepatomegaly, and liver dysfunction, hypotonia, muscle weakness, exercise intolerance, developmental delay, delayed motor development, mental retardation, and lactic acidosis. Some affected individuals manifest fatal hypertrophic cardiomyopathy resulting in neonatal death. A subset of patients also suffers from Leigh syndrome.[13][14][8][9] Specifically, cases of pathogenic SCO1 mutations have resulted in fatal infantile encephalopathy, neonatal-onset hepatic failure, and severe hepatopathy. The P174L and M294V mutations have been identified and implicated in these diseases and phenotypes.[14][15][16] It has also been suggested that mutations in SCO1, as well as SCO2, can result in a cellular copper deficiency, which can occur separately from cytochrome c oxidase assembly defects.[13]
Model organisms
| Characteristic | Phenotype |
|---|---|
| Homozygote viability | Abnormal |
| Recessive lethal study | Abnormal |
| Fertility | Normal |
| Body weight | Normal |
| Anxiety | Normal |
| Neurological assessment | Normal |
| Grip strength | Normal |
| Hot plate | Normal |
| Dysmorphology | Normal |
| Indirect calorimetry | Normal |
| Glucose tolerance test | Normal |
| Auditory brainstem response | Normal |
| DEXA | Normal |
| Radiography | Normal |
| Body temperature | Normal |
| Eye morphology | Normal |
| Clinical chemistry | Normal |
| Haematology | Normal |
| Peripheral blood lymphocytes | Normal |
| Micronucleus test | Normal |
| Heart weight | Normal |
| Salmonella infection | Normal[17] |
| All tests and analysis from[18][19] |
Model organisms have been used in the study of SCO1 function. A conditional knockout mouse line, called Sco1tm1a(KOMP)Wtsi[20][21] was generated as part of the International Knockout Mouse Consortium program—a high-throughput mutagenesis project to generate and distribute animal models of disease.[22][23][24]
Male and female animals underwent a standardized phenotypic screen to determine the effects of deletion.[18][25] Twenty-two tests were carried out on mutant mice and two significant abnormalities were observed.[18] No homozygous mutant embryos were identified during gestation, and therefore none survived until weaning. The remaining tests were carried out on heterozygous mutant adult mice; no additional significant abnormalities were observed in these animals.[18]
Interactions
SCO1 has been shown to have 127 binary protein-protein interactions including 120 co-complex interactions. SCO1 interacts with COA6, TMEM177, COX20, COX16, COX17, WDR19, CIDEB, and UBC7. It is also found in a complex with TMEM177, COX20, COA6, MT-CO2/COX2, COX18, and SCO2.[26][8][9][27]
References
- GRCh38: Ensembl release 89: ENSG00000133028 - Ensembl, May 2017
- GRCm38: Ensembl release 89: ENSMUSG00000069844 - Ensembl, May 2017
- "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- Petruzzella V, Tiranti V, Fernandez P, Ianna P, Carrozzo R, Zeviani M (December 1998). "Identification and characterization of human cDNAs specific to BCS1, PET112, SCO1, COX15, and COX11, five genes involved in the formation and function of the mitochondrial respiratory chain". Genomics. 54 (3): 494–504. doi:10.1006/geno.1998.5580. PMID 9878253.
- "Entrez Gene: SCO1 SCO cytochrome oxidase deficient homolog 1 (yeast)".
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - Brosel S, Yang H, Tanji K, Bonilla E, Schon EA (November 2010). "Unexpected vascular enrichment of SCO1 over SCO2 in mammalian tissues: implications for human mitochondrial disease". The American Journal of Pathology. 177 (5): 2541–8. doi:10.2353/ajpath.2010.100229. PMC 2966810. PMID 20864674.
- "UniProt: the universal protein knowledgebase". Nucleic Acids Research. 45 (D1): D158–D169. January 2017. doi:10.1093/nar/gkw1099. PMC 5210571. PMID 27899622.
- "SCO1 - Protein SCO1 homolog, mitochondrial precursor - Homo sapiens (Human) - SCO1 gene & protein". www.uniprot.org. Retrieved 2018-08-08.
 This article incorporates text available under the CC BY 4.0 license.
This article incorporates text available under the CC BY 4.0 license. - Yao, Daniel. "Cardiac Organellar Protein Atlas Knowledgebase (COPaKB) —— Protein Information". amino.heartproteome.org. Retrieved 2018-08-08.
- Zong NC, Li H, Li H, Lam MP, Jimenez RC, Kim CS, et al. (October 2013). "Integration of cardiac proteome biology and medicine by a specialized knowledgebase". Circulation Research. 113 (9): 1043–53. doi:10.1161/CIRCRESAHA.113.301151. PMC 4076475. PMID 23965338.
- Leary SC, Kaufman BA, Pellecchia G, Guercin GH, Mattman A, Jaksch M, Shoubridge EA (September 2004). "Human SCO1 and SCO2 have independent, cooperative functions in copper delivery to cytochrome c oxidase". Human Molecular Genetics. 13 (17): 1839–48. doi:10.1093/hmg/ddh197. PMID 15229189.
- Leary SC, Cobine PA, Kaufman BA, Guercin GH, Mattman A, Palaty J, Lockitch G, Winge DR, Rustin P, Horvath R, Shoubridge EA (January 2007). "The human cytochrome c oxidase assembly factors SCO1 and SCO2 have regulatory roles in the maintenance of cellular copper homeostasis". Cell Metabolism. 5 (1): 9–20. doi:10.1016/j.cmet.2006.12.001. PMID 17189203.
- Valnot I, Osmond S, Gigarel N, Mehaye B, Amiel J, Cormier-Daire V, Munnich A, Bonnefont JP, Rustin P, Rötig A (November 2000). "Mutations of the SCO1 gene in mitochondrial cytochrome c oxidase deficiency with neonatal-onset hepatic failure and encephalopathy". American Journal of Human Genetics. 67 (5): 1104–9. doi:10.1016/S0002-9297(07)62940-1. PMC 1288552. PMID 11013136.
- Banci L, Bertini I, Ciofi-Baffoni S, Leontari I, Martinelli M, Palumaa P, Sillard R, Wang S (January 2007). "Human Sco1 functional studies and pathological implications of the P174L mutant". Proceedings of the National Academy of Sciences of the United States of America. 104 (1): 15–20. doi:10.1073/pnas.0606189103. PMC 1765425. PMID 17182746.
- Leary SC, Antonicka H, Sasarman F, Weraarpachai W, Cobine PA, Pan M, Brown GK, Brown R, Majewski J, Ha KC, Rahman S, Shoubridge EA (October 2013). "Novel mutations in SCO1 as a cause of fatal infantile encephalopathy and lactic acidosis". Human Mutation. 34 (10): 1366–70. doi:10.1002/humu.22385. PMID 23878101. S2CID 43630957.
- "Salmonella infection data for Sco1". Wellcome Trust Sanger Institute.
- Gerdin, AK (2010). "The Sanger Mouse Genetics Programme: High throughput characterisation of knockout mice". Acta Ophthalmologica. 88: 925–7. doi:10.1111/j.1755-3768.2010.4142.x. S2CID 85911512.
- Mouse Resources Portal, Wellcome Trust Sanger Institute.
- "International Knockout Mouse Consortium".
- "Mouse Genome Informatics".
- Skarnes WC, Rosen B, West AP, Koutsourakis M, Bushell W, Iyer V, et al. (June 2011). "A conditional knockout resource for the genome-wide study of mouse gene function". Nature. 474 (7351): 337–42. doi:10.1038/nature10163. PMC 3572410. PMID 21677750.
- Dolgin E (June 2011). "Mouse library set to be knockout". Nature. 474 (7351): 262–3. doi:10.1038/474262a. PMID 21677718.
- Collins FS, Rossant J, Wurst W (January 2007). "A mouse for all reasons". Cell. 128 (1): 9–13. doi:10.1016/j.cell.2006.12.018. PMID 17218247. S2CID 18872015.
- van der Weyden L, White JK, Adams DJ, Logan DW (June 2011). "The mouse genetics toolkit: revealing function and mechanism". Genome Biology. 12 (6): 224. doi:10.1186/gb-2011-12-6-224. PMC 3218837. PMID 21722353.
- Lorenzi I, Oeljeklaus S, Aich A, Ronsör C, Callegari S, Dudek J, Warscheid B, Dennerlein S, Rehling P (February 2018). "The mitochondrial TMEM177 associates with COX20 during COX2 biogenesis". Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 1865 (2): 323–333. doi:10.1016/j.bbamcr.2017.11.010. PMC 5764226. PMID 29154948.
- "127 binary interactions found for search term SCO1". IntAct Molecular Interaction Database. EMBL-EBI. Retrieved 2018-08-25.
Further reading
- Shoubridge EA (2001). "Cytochrome c oxidase deficiency". American Journal of Medical Genetics. 106 (1): 46–52. doi:10.1002/ajmg.1378. PMID 11579424.
- Schulze M, Rödel G (March 1989). "Accumulation of the cytochrome c oxidase subunits I and II in yeast requires a mitochondrial membrane-associated protein, encoded by the nuclear SCO1 gene". Molecular & General Genetics. 216 (1): 37–43. doi:10.1007/BF00332228. PMID 2543907. S2CID 13029649.
- Schulze M, Rödel G (March 1988). "SCO1, a yeast nuclear gene essential for accumulation of mitochondrial cytochrome c oxidase subunit II". Molecular & General Genetics. 211 (3): 492–8. doi:10.1007/BF00425706. PMID 2835635. S2CID 25345429.
- Andersson B, Wentland MA, Ricafrente JY, Liu W, Gibbs RA (April 1996). "A "double adaptor" method for improved shotgun library construction". Analytical Biochemistry. 236 (1): 107–13. doi:10.1006/abio.1996.0138. PMID 8619474.
- Yu W, Andersson B, Worley KC, Muzny DM, Ding Y, Liu W, Ricafrente JY, Wentland MA, Lennon G, Gibbs RA (April 1997). "Large-scale concatenation cDNA sequencing". Genome Research. 7 (4): 353–8. doi:10.1101/gr.7.4.353. PMC 139146. PMID 9110174.
- Paret C, Ostermann K, Krause-Buchholz U, Rentzsch A, Rödel G (March 1999). "Human members of the SCO1 gene family: complementation analysis in yeast and intracellular localization". FEBS Letters. 447 (1): 65–70. doi:10.1016/S0014-5793(99)00266-5. PMID 10218584. S2CID 7599827.
- Papadopoulou LC, Sue CM, Davidson MM, Tanji K, Nishino I, Sadlock JE, Krishna S, Walker W, Selby J, Glerum DM, Coster RV, Lyon G, Scalais E, Lebel R, Kaplan P, Shanske S, De Vivo DC, Bonilla E, Hirano M, DiMauro S, Schon EA (November 1999). "Fatal infantile cardioencephalomyopathy with COX deficiency and mutations in SCO2, a COX assembly gene". Nature Genetics. 23 (3): 333–7. doi:10.1038/15513. PMID 10545952. S2CID 23387553.
- Horvath R, Lochmüller H, Stucka R, Yao J, Shoubridge EA, Kim SH, Gerbitz KD, Jaksch M (September 2000). "Characterization of human SCO1 and COX17 genes in mitochondrial cytochrome-c-oxidase deficiency". Biochemical and Biophysical Research Communications. 276 (2): 530–3. doi:10.1006/bbrc.2000.3495. PMID 11027508.
- Williams JC, Sue C, Banting GS, Yang H, Glerum DM, Hendrickson WA, Schon EA (April 2005). "Crystal structure of human SCO1: implications for redox signaling by a mitochondrial cytochrome c oxidase "assembly" protein". The Journal of Biological Chemistry. 280 (15): 15202–11. doi:10.1074/jbc.M410705200. PMID 15659396.
- Horng YC, Leary SC, Cobine PA, Young FB, George GN, Shoubridge EA, Winge DR (October 2005). "Human Sco1 and Sco2 function as copper-binding proteins". The Journal of Biological Chemistry. 280 (40): 34113–22. doi:10.1074/jbc.M506801200. PMID 16091356.
- Cobine PA, Pierrel F, Leary SC, Sasarman F, Horng YC, Shoubridge EA, Winge DR (May 2006). "The P174L mutation in human Sco1 severely compromises Cox17-dependent metallation but does not impair copper binding". The Journal of Biological Chemistry. 281 (18): 12270–6. doi:10.1074/jbc.M600496200. PMID 16520371.
- Banci L, Bertini I, Calderone V, Ciofi-Baffoni S, Mangani S, Martinelli M, Palumaa P, Wang S (June 2006). "A hint for the function of human Sco1 from different structures". Proceedings of the National Academy of Sciences of the United States of America. 103 (23): 8595–600. doi:10.1073/pnas.0601375103. PMC 1482625. PMID 16735468.
- Leary SC, Sasarman F, Nishimura T, Shoubridge EA (June 2009). "Human SCO2 is required for the synthesis of CO II and as a thiol-disulphide oxidoreductase for SCO1". Human Molecular Genetics. 18 (12): 2230–40. doi:10.1093/hmg/ddp158. PMID 19336478.
- Stiburek L, Vesela K, Hansikova H, Hulkova H, Zeman J (May 2009). "Loss of function of Sco1 and its interaction with cytochrome c oxidase". American Journal of Physiology. Cell Physiology. 296 (5): C1218–26. doi:10.1152/ajpcell.00564.2008. PMID 19295170.
PDB gallery | |
|---|---|
|
This article incorporates text from the United States National Library of Medicine, which is in the public domain.