Cytochrome c oxidase subunit II
Cytochrome c oxidase subunit 2, also known as cytochrome c oxidase polypeptide II, is a protein that in humans is encoded by the MT-CO2 gene.[4] Cytochrome c oxidase subunit II, abbreviated COXII, COX2, COII, or MT-CO2, is the second subunit of cytochrome c oxidase. It is also one of the three mitochondrial DNA (mtDNA) encoded subunits (MT-CO1, MT-CO2, MT-CO3) of respiratory complex IV.
| Cytochrome c oxidase subunit II, transmembrane domain | |||||||||
|---|---|---|---|---|---|---|---|---|---|
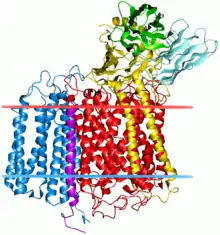 Bacterial cytochrome c oxidase complex. Subunit II indicated by blue. | |||||||||
| Identifiers | |||||||||
| Symbol | COX2_TM | ||||||||
| Pfam | PF02790 | ||||||||
| InterPro | IPR011759 | ||||||||
| PROSITE | PDOC00075 | ||||||||
| SCOP2 | 1occ / SCOPe / SUPFAM | ||||||||
| TCDB | 3.D.4 | ||||||||
| OPM superfamily | 4 | ||||||||
| OPM protein | 1v55 | ||||||||
| |||||||||
| Cytochrome C oxidase subunit II, periplasmic domain | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Identifiers | |||||||||
| Symbol | COX2 | ||||||||
| Pfam | PF00116 | ||||||||
| InterPro | IPR002429 | ||||||||
| CDD | cd13912 | ||||||||
| |||||||||
Structure
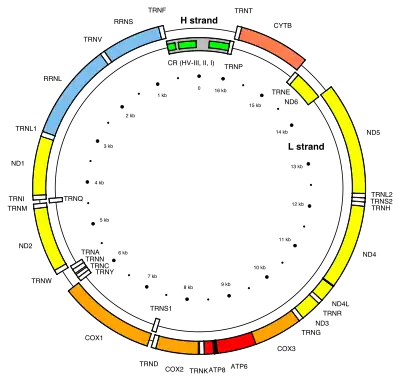
The MT-CO2 gene is located on the p arm of mitochondrial DNA at position 12 and it spans 683 base pairs.[4] The MT-CO2 gene produces a 25.6 kDa protein composed of 227 amino acids.[5][6] MT-CO2 is a subunit of the enzyme Cytochrome c oxidase (EC 1.9.3.1)[7][8] (Complex IV), an oligomeric enzymatic complex of the mitochondrial respiratory chain involved in the transfer of electrons from cytochrome c to oxygen. In eukaryotes this enzyme complex is located in the mitochondrial inner membrane; in aerobic prokaryotes it is found in the plasma membrane. The enzyme complex consists of 3-4 subunits (prokaryotes) to up to 13 polypeptides (mammals). The N-terminal domain of cytochrome C oxidase contains two transmembrane alpha-helices.[8][7] The structure of MT-CO2 is known to contain one redox center and a binuclear copper A center (CuA). The CuA is located in a conserved cysteine loop at 196 and 200 amino acid positions and conserved histidine at 204. Several bacterial MT-CO2 have a C-terminal extension that contains a covalently bound haem c.[9][10]
Function
The MT-CO2 gene encodes for the second subunit of cytochrome c oxidase (complex IV), a component of the mitochondrial respiratory chain that catalyzes the reduction of oxygen to water. MT-CO2 is one of the three subunits which are responsible for the formation of the functional core of the cytochrome c oxidase. MT-CO2 plays an essential role in the transfer of electrons from cytochrome c to the bimetallic center of the catalytic subunit 1 by utilizing its binuclear copper A center. It contains two adjacent transmembrane regions in its N-terminus and the major part of the protein is exposed to the periplasmic or to the mitochondrial intermembrane space, respectively. MT-CO2 provides the substrate-binding site and contains the binuclear copper A center, probably the primary acceptor in cytochrome c oxidase.[11][12][4]
Clinical significance
Mitochondrial complex IV deficiency
Variants of MT-CO2 have been associated with the mitochondrial Complex IV deficiency, a deficiency in an enzyme complex of the mitochondrial respiratory chain that catalyzes the oxidation of cytochrome c utilizing molecular oxygen.[13] The deficiency is characterized by heterogeneous phenotypes ranging from isolated myopathy to severe multisystem disease affecting several tissues and organs. Other Clinical Manifestations include hypertrophic cardiomyopathy, hepatomegaly and liver dysfunction, hypotonia, muscle weakness, exercise intolerance, developmental delay, delayed motor development and mental retardation.[14] Mutations of MT-CO2 is also known to cause Leigh's disease, which may be caused by an abnormality or deficiency of cytochrome oxidase.[8][7]
A wide range of symptoms have been found in patients with pathogenic mutations in the MT-CO2 gene with the mitochonrdial Complex IV deficiency. A deletion mutation of a single nucleotide (7630delT) in the gene has been found to cause symptoms of reversible aphasia, right hemiparesis, hemianopsia, exercise intolerance, progressive mental impairment, and short stature.[15] Furthermore, a patient with a nonsense mutation (7896G>A) of the gene resulted in phenotypes such as short stature, low weight, microcephaly, skin abnormalities, severe hypotonia, and normal reflexes.[16] A novel heteroplasmic mutation (7587T>C) which altered the initiation codon of the MT-CO2 gene in patients have shown clinical manifestations such as progressive gait ataxia, cognitive impairment, bilateral optic atrophy, pigmentary retinopathy, a decrease in color vision, and mild distal-muscle wasting.[17]
Others
Juvenile myopathy, encephalopathy, lactic acidosis, and stroke have also been associated with mutations in the MT-CO2 gene.[4]
Interactions
MT-CO2 is known to interact with cytochrome c by the utilization of a lysine ring around the carboxyl containing heme edge of cytochrome c in MT-CO2, including glutamate 129, aspartate 132, and glutamate 19.
References
- GRCm38: Ensembl release 89: ENSMUSG00000064354 - Ensembl, May 2017
- "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Entrez Gene: COX2 cytochrome c oxidase subunit II".
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - Zong NC, Li H, Li H, Lam MP, Jimenez RC, Kim CS, Deng N, Kim AK, Choi JH, Zelaya I, Liem D, Meyer D, Odeberg J, Fang C, Lu HJ, Xu T, Weiss J, Duan H, Uhlen M, Yates JR, Apweiler R, Ge J, Hermjakob H, Ping P (October 2013). "Integration of cardiac proteome biology and medicine by a specialized knowledgebase". Circulation Research. 113 (9): 1043–53. doi:10.1161/CIRCRESAHA.113.301151. PMC 4076475. PMID 23965338.
- "Cytochrome c oxidase subunit 2". Cardiac Organellar Protein Atlas Knowledgebase (COPaKB).
- Capaldi RA, Malatesta F, Darley-Usmar VM (July 1983). "Structure of cytochrome c oxidase". Biochimica et Biophysica Acta (BBA) - Reviews on Bioenergetics. 726 (2): 135–48. doi:10.1016/0304-4173(83)90003-4. PMID 6307356.
- García-Horsman JA, Barquera B, Rumbley J, Ma J, Gennis RB (September 1994). "The superfamily of heme-copper respiratory oxidases". Journal of Bacteriology. 176 (18): 5587–600. doi:10.1128/jb.176.18.5587-5600.1994. PMC 196760. PMID 8083153.
- Capaldi RA (1990). "Structure and function of cytochrome c oxidase". Annual Review of Biochemistry. 59: 569–96. doi:10.1146/annurev.bi.59.070190.003033. PMID 2165384.
- Hill BC (April 1993). "The sequence of electron carriers in the reaction of cytochrome c oxidase with oxygen". Journal of Bioenergetics and Biomembranes. 25 (2): 115–20. doi:10.1007/bf00762853. PMID 8389744. S2CID 45975377.
-
"MT-CO2 - Cytochrome c oxidase subunit 2 - Homo sapiens (Human) - MT-CO2 gene & protein". Retrieved 2018-08-07.
 This article incorporates text available under the CC BY 4.0 license.
This article incorporates text available under the CC BY 4.0 license.
- "UniProt: the universal protein knowledgebase". Nucleic Acids Research. 45 (D1): D158–D169. January 2017. doi:10.1093/nar/gkw1099. PMC 5210571. PMID 27899622.
- Ostergaard E, Weraarpachai W, Ravn K, Born AP, Jønson L, Duno M, Wibrand F, Shoubridge EA, Vissing J (March 2015). "Mutations in COA3 cause isolated complex IV deficiency associated with neuropathy, exercise intolerance, obesity, and short stature". Journal of Medical Genetics. 52 (3): 203–7. doi:10.1136/jmedgenet-2014-102914. PMID 25604084. S2CID 43018915.
- "Mitochondrial complex IV deficiency". www.uniprot.org.
- Rossmanith W, Freilinger M, Roka J, Raffelsberger T, Moser-Thier K, Prayer D, Bernert G, Bittner RE (February 2008). "Isolated cytochrome c oxidase deficiency as a cause of MELAS". Journal of Medical Genetics. 45 (2): 117–21. doi:10.1136/jmg.2007.052076. PMC 3027970. PMID 18245391.
- Campos Y, García-Redondo A, Fernández-Moreno MA, Martínez-Pardo M, Goda G, Rubio JC, Martín MA, del Hoyo P, Cabello A, Bornstein B, Garesse R, Arenas J (September 2001). "Early-onset multisystem mitochondrial disorder caused by a nonsense mutation in the mitochondrial DNA cytochrome C oxidase II gene". Annals of Neurology. 50 (3): 409–13. doi:10.1002/ana.1141. PMID 11558799. S2CID 23891106.
- Clark KM, Taylor RW, Johnson MA, Chinnery PF, Chrzanowska-Lightowlers ZM, Andrews RM, Nelson IP, Wood NW, Lamont PJ, Hanna MG, Lightowlers RN, Turnbull DM (May 1999). "An mtDNA mutation in the initiation codon of the cytochrome C oxidase subunit II gene results in lower levels of the protein and a mitochondrial encephalomyopathy". American Journal of Human Genetics. 64 (5): 1330–9. doi:10.1086/302361. PMC 1377869. PMID 10205264.
Further reading
- Torroni A, Achilli A, Macaulay V, Richards M, Bandelt HJ (June 2006). "Harvesting the fruit of the human mtDNA tree". Trends in Genetics. 22 (6): 339–45. doi:10.1016/j.tig.2006.04.001. PMID 16678300.
- Barrell BG, Bankier AT, Drouin J (November 1979). "A different genetic code in human mitochondria". Nature. 282 (5735): 189–94. Bibcode:1979Natur.282..189B. doi:10.1038/282189a0. PMID 226894. S2CID 4335828.
- Bodenteich A, Mitchell LG, Polymeropoulos MH, Merril CR (May 1992). "Dinucleotide repeat in the human mitochondrial D-loop". Human Molecular Genetics. 1 (2): 140. doi:10.1093/hmg/1.2.140-a. PMID 1301157.
- Lu X, Walker T, MacManus JP, Seligy VL (July 1992). "Differentiation of HT-29 human colonic adenocarcinoma cells correlates with increased expression of mitochondrial RNA: effects of trehalose on cell growth and maturation". Cancer Research. 52 (13): 3718–25. PMID 1377597.
- Marzuki S, Noer AS, Lertrit P, Thyagarajan D, Kapsa R, Utthanaphol P, Byrne E (December 1991). "Normal variants of human mitochondrial DNA and translation products: the building of a reference data base". Human Genetics. 88 (2): 139–45. doi:10.1007/bf00206061. PMID 1757091. S2CID 28048453.
- Moraes CT, Andreetta F, Bonilla E, Shanske S, DiMauro S, Schon EA (March 1991). "Replication-competent human mitochondrial DNA lacking the heavy-strand promoter region". Molecular and Cellular Biology. 11 (3): 1631–7. doi:10.1128/MCB.11.3.1631. PMC 369459. PMID 1996112.
- Power MD, Kiefer MC, Barr PJ, Reeves R (August 1989). "Nucleotide sequence of human mitochondrial cytochrome c oxidase II cDNA". Nucleic Acids Research. 17 (16): 6734. doi:10.1093/nar/17.16.6734. PMC 318375. PMID 2550900.
- Attardi G, Chomyn A, Doolittle RF, Mariottini P, Ragan CI (1987). "Seven unidentified reading frames of human mitochondrial DNA encode subunits of the respiratory chain NADH dehydrogenase". Cold Spring Harbor Symposia on Quantitative Biology. 51 (1): 103–14. doi:10.1101/sqb.1986.051.01.013. PMID 3472707.
- Chomyn A, Cleeter MW, Ragan CI, Riley M, Doolittle RF, Attardi G (October 1986). "URF6, last unidentified reading frame of human mtDNA, codes for an NADH dehydrogenase subunit". Science. 234 (4776): 614–8. Bibcode:1986Sci...234..614C. doi:10.1126/science.3764430. PMID 3764430.
- Chomyn A, Mariottini P, Cleeter MW, Ragan CI, Matsuno-Yagi A, Hatefi Y, Doolittle RF, Attardi G (1985). "Six unidentified reading frames of human mitochondrial DNA encode components of the respiratory-chain NADH dehydrogenase". Nature. 314 (6012): 592–7. Bibcode:1985Natur.314..592C. doi:10.1038/314592a0. PMID 3921850. S2CID 32964006.
- Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, Staden R, Young IG (April 1981). "Sequence and organization of the human mitochondrial genome". Nature. 290 (5806): 457–65. Bibcode:1981Natur.290..457A. doi:10.1038/290457a0. PMID 7219534. S2CID 4355527.
- Montoya J, Ojala D, Attardi G (April 1981). "Distinctive features of the 5'-terminal sequences of the human mitochondrial mRNAs". Nature. 290 (5806): 465–70. Bibcode:1981Natur.290..465M. doi:10.1038/290465a0. PMID 7219535. S2CID 4358928.
- Horai S, Hayasaka K, Kondo R, Tsugane K, Takahata N (January 1995). "Recent African origin of modern humans revealed by complete sequences of hominoid mitochondrial DNAs". Proceedings of the National Academy of Sciences of the United States of America. 92 (2): 532–6. Bibcode:1995PNAS...92..532H. doi:10.1073/pnas.92.2.532. PMC 42775. PMID 7530363.
- Ruvolo M, Zehr S, von Dornum M, Pan D, Chang B, Lin J (November 1993). "Mitochondrial COII sequences and modern human origins". Molecular Biology and Evolution. 10 (6): 1115–35. doi:10.1093/oxfordjournals.molbev.a040068. PMID 8277847.
- Polyak K, Li Y, Zhu H, Lengauer C, Willson JK, Markowitz SD, Trush MA, Kinzler KW, Vogelstein B (November 1998). "Somatic mutations of the mitochondrial genome in human colorectal tumours". Nature Genetics. 20 (3): 291–3. doi:10.1038/3108. PMID 9806551. S2CID 19786796.
- Kato MV (March 1999). "The mechanisms of death of an erythroleukemic cell line by p53: involvement of the microtubule and mitochondria". Leukemia & Lymphoma. 33 (1–2): 181–6. doi:10.3109/10428199909093740. PMID 10194136.
- Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N (October 1999). "Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA". Nature Genetics. 23 (2): 147. doi:10.1038/13779. PMID 10508508. S2CID 32212178.
- Ingman M, Kaessmann H, Pääbo S, Gyllensten U (December 2000). "Mitochondrial genome variation and the origin of modern humans". Nature. 408 (6813): 708–13. Bibcode:2000Natur.408..708I. doi:10.1038/35047064. PMID 11130070. S2CID 52850476.
- Finnilä S, Lehtonen MS, Majamaa K (June 2001). "Phylogenetic network for European mtDNA". American Journal of Human Genetics. 68 (6): 1475–84. doi:10.1086/320591. PMC 1226134. PMID 11349229.
- Maca-Meyer N, González AM, Larruga JM, Flores C, Cabrera VM (2003). "Major genomic mitochondrial lineages delineate early human expansions". BMC Genetics. 2: 13. doi:10.1186/1471-2156-2-13. PMC 55343. PMID 11553319.
This article incorporates text from the United States National Library of Medicine, which is in the public domain.