Silent mutation
Silent mutations are mutations in DNA that do not have an observable effect on the organism's phenotype. They are a specific type of neutral mutation. The phrase silent mutation is often used interchangeably with the phrase synonymous mutation; however, synonymous mutations are not always silent, nor vice versa.[1][2][3][4][5] Synonymous mutations can affect transcription, splicing, mRNA transport, and translation, any of which could alter phenotype, rendering the synonymous mutation non-silent.[3] The substrate specificity of the tRNA to the rare codon can affect the timing of translation, and in turn the co-translational folding of the protein.[1] This is reflected in the codon usage bias that is observed in many species. Mutations that cause the altered codon to produce an amino acid with similar functionality (e.g. a mutation producing leucine instead of isoleucine) are often classified as silent; if the properties of the amino acid are conserved, this mutation does not usually significantly affect protein function.[6]
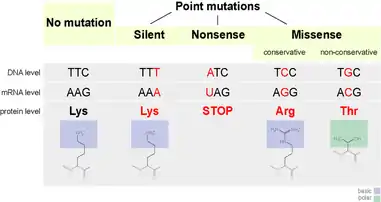
Genetic code
The genetic code translates mRNA nucleotide sequences to amino acid sequences. Genetic information is coded using this process with groups of three nucleotides along the mRNA which are commonly known as codons.[7] The set of three nucleotides almost always produce the same amino acid with are a few exceptions like UGA which typically serves as the stop codon but can also encode tryptophan in mammalian mitochondria.[7] Most amino acids are specified by multiple codons demonstrating that the genetic code is degenerate–different codons result in the same amino acid.[7] Codons that code for the same amino acid are termed synonyms. Silent mutations are base substitutions that result in no change of the amino acid or amino acid functionality when the altered messenger RNA (mRNA) is translated. For example, if the codon AAA is altered to become AAG, the same amino acid – lysine – will be incorporated into the peptide chain.
Mutations are often linked to diseases or negative impacts but silent mutations can be extremely beneficial in creating genetic diversity among species in a population. Germ-line mutations are passed from the parent to the offspring.[8] Scientists have predicted that people have approximately 5 to 10 deadly mutations in their genomes but this is essentially harmless because there is usually only one copy of a particular bad gene so diseases are unlikely.[8] Silent mutations can also be produced by insertions or deletions, which cause a shift in the reading frame.[9]
Because silent mutations do not alter protein function they are often treated as though they are evolutionarily neutral. Many organisms are known to exhibit codon usage biases, suggesting that there is selection for the use of particular codons due to the need for translational stability. Transfer RNA (tRNA) availability is one of the reasons that silent mutations might not be as silent as conventionally believed.[10]
There is a different tRNA molecule for each codon. For example, there is a specific tRNA molecule for the codon UCU and another specific for the codon UCC, both of which code for the amino acid serine. In this instance, if there was a thousand times less UCC tRNA than UCU tRNA, then the incorporation of serine into a polypeptide chain would happen a thousand times more slowly when a mutation causes the codon to change from UCU to UCC. If amino acid transport to the ribosome is delayed, translation will be carried out at a much slower rate. This can result in lower expression of a particular gene containing that silent mutation if the mutation occurs within an exon. Additionally, if the ribosome has to wait too long to receive the amino acid, the ribosome could terminate translation prematurely.[6]
Structural consequences
Primary structure
A nonsynonymous mutation that occurs at the genomic or transcriptional levels is one that results in an alteration to the amino acid sequence in the protein product. A protein's primary structure refers to its amino acid sequence. A substitution of one amino acid for another can impair protein function and tertiary structure, however its effects may be minimal or tolerated depending on how closely the properties of the amino acids involved in the swap correlate.[11] The premature insertion of a stop codon, a nonsense mutation, can alter the primary structure of a protein.[12] In this case, a truncated protein is produced. Protein function and folding is dependent on the position in which the stop codon was inserted and the amount and composition of the sequence lost.
Conversely, silent mutations are mutations in which the amino acid sequence is not altered.[12] Silent mutations lead to a change of one of the letters in the triplet code that represents a codon, but despite the single base change, the amino acid that is coded for remains unchanged or similar in biochemical properties. This is permitted by the degeneracy of the genetic code.
Historically, silent mutations were thought to be of little to no significance. However, recent research suggests that such alterations to the triplet code do affect protein translation efficiency and protein folding and function.[13][14]
Furthermore, a change in primary structure is critical because the fully folded tertiary structure of a protein is dependent upon the primary structure. The discovery was made throughout a series of experiments in the 1960s that discovered that reduced and denatured RNase in its unfolded form could refold into the native tertiary form. The tertiary structure of a protein is a fully folded polypeptide chain with all hydrophobic R-groups folded into the interior of the protein to maximize entropy with interactions between secondary structures such as beta sheets and alpha helixes. Since the structure of proteins determines its function, it is critical that a protein be folded correctly into its tertiary form so that the protein will function properly. However, it is important to note that polypeptide chains may differ vastly in primary structure, but be very similar in tertiary structure and protein function.[15]
Secondary structure
Silent mutations alter the secondary structure of mRNA.
Secondary structure of proteins consists of interactions between the atoms of the backbone of a polypeptide chain, excluding the R-groups. One common type of secondary structures is the alpha helix, which is a right-handed helix that results from hydrogen bonds between the nth amino acid residue and the n+4th amino acid residue. The other common type of secondary structure is the beta sheet, which displays a right-handed twist, can be parallel or anti-parallel depending on the direction of the direction of the bonded polypeptides, and consists of hydrogen bonds between the carbonyl and amino groups of the backbone of two polypeptide chains.[16]
mRNA has a secondary structure that is not necessarily linear like that of DNA, thus the shape that accompanies complementary bonding in the structure can have significant effects. For example, if the mRNA molecule is relatively unstable, then it can be rapidly degraded by enzymes in the cytoplasm. If the RNA molecule is highly stable, and the complementary bonds are strong and resistant to unpacking prior to translation, then the gene may be under expressed. Codon usage influences mRNA stability.[10]
Furthermore, since all organisms contain a slightly different genetic code, their mRNA structures differ slightly as well, however, multiple studies have been conducted that show that all properly folded mRNA structures are dependent on the primary sequence of the polypeptide chain and that the structure is maintained by dinucleotide relative abundances in the cell matrix. It has also been discovered that mRNA secondary structure is important for cell processes such as transcript stability and translation. The general idea is that the functional domains of mRNA fold upon each other, while the start and stop codon regions generally are more relaxed, which could aid in the signaling of initiation and termination in translation.[17]
If the oncoming ribosome pauses because of a knot in the RNA, then the polypeptide could potentially have enough time to fold into a non-native structure before the tRNA molecule can add another amino acid. Silent mutations may also affect splicing, or transcriptional control.
Tertiary structure
Silent mutations affect protein folding and function.[1] Normally a misfolded protein can be refolded with the help of molecular chaperones. RNA typically produces two common misfolded proteins by tending to fold together and become stuck in different conformations and it has a difficulty singling in on the favored specific tertiary structure because of other competing structures. RNA-binding proteins can assist RNA folding problems, however, when a silent mutation occurs in the mRNA chain, these chaperones do not bind properly to the molecule and are unable to redirect the mRNA into the correct fold.[18]
Recent research suggests that silent mutations can have an effect on subsequent protein structure and activity.[19][20] The timing and rate of protein folding can be altered, which can lead to functional impairments.[21]
Research and clinical applications
Silent mutations have been employed as an experimental strategy and can have clinical implications.
Steffen Mueller at the Stony Brook University designed a live vaccine for polio in which the virus was engineered to have synonymous codons replace naturally occurring ones in the genome. As a result, the virus was still able to infect and reproduce, albeit more slowly. Mice that were vaccinated with this vaccine and exhibited resistance against the natural polio strain.
In molecular cloning experiments, it can be useful to introduce silent mutations into a gene of interest in order to create or remove recognition sites for restriction enzymes.
Mental disorders can be caused by silent mutations. One silent mutation causes the dopamine receptor D2 gene to be less stable and degrade faster, underexpressing the gene.
A silent mutation in the multidrug resistance gene 1 (MDR1), which codes for a cellular membrane pump that expels drugs from the cell, can slow down translation in a specific location to allow the peptide chain to bend into an unusual conformation. Thus, the mutant pump is less functional.
Deviations from average pain sensitivity are caused by both an ATG to GTG mutation (nonsynonymous), and a CAT to CAC mutation (synonymous). These two mutations are both shared by the low pain sensitivity and high pain sensitivity gene. Low pain sensitivity has an additional CTC to CTG silent mutation, while high pain sensitivity does not and shares the CTC sequence at this location with average pain sensitivity.[22]
| LPS | APS | HPS |
|---|---|---|
| CAC | CAT | CAC |
| CTG | CTC | CTC |
| GTG | ATG | GTG |
Multi-Drug Resistance Gene 1
Around 99.8% of genes that undergo mutations are deemed silent because the nucleotide change does not change the amino acid being translated.[23] Although silent mutations are not supposed to have an effect on the phenotypic outcome, some mutations prove otherwise like the Multi-Drug Resistance Gene 1. MDR1 codes for the P-glycoprotein which helps get rid of drugs in the body. It is located in the intestines, liver, pancreas, and brain. MDR 1 is located in the same places that CYP3A4 is located in, which is an enzyme that helps get rid of toxins or drugs from the liver and intestines. Silent mutations like MDR 1 do express a change in phenotypic response. A study done on mice showed when they did not have enough of the MDR 1 gene, their body did not recognize the ivermectin or cyclosporine drug, leading to the creation of toxins in their bodies.[23]
MRD1 has over fifty single nucleotide polymorphisms (SNP's) which are changes in the nucleotide base sequence.[24][23] In MDR1 the gene exon 26 which represents 3535C can mutate to 3535T which then changes the transfer RNA into one that is not often as seen, leading to changes in the outcome during translation. This is an example of how some silent mutations are not always silent.[25] The multi-drug resistance genes at Exon 26 C3435T, exon 21 G2677T/A, and exon 12 C1236T have been studied to have SNP's that occur at the same time, therefore making the phenotypic "function "change. This suggests a haplotype dependency between exon 26 and other exon that have polymorphisms. For example, efavirenz and nelfinavir are two types of drugs that help decrease the HIV infection in a person's body. When the SNP from exon 26 is coupled with other SNP exons, the drugs have a lower chance of maintaining the HIV infection. Although, when the TT nucleotides in exon 26 are expressed the patient has a lower concentration of the virus but when the genotype morphs into CC or CT the infection is able to spread like normal leaving the MDR 1 gene almost defenseless. These changes in bases of exon 26 for MDR 1 show a correlation between the MDR 1 gene mutations and the ability of the antiretroviral drugs to suppress the HIV infection.[23]
Exon 26 has also been studied as to whether it is haplotype dependent or not. The presence of the SNP of exon 26 changes phenotypic functions when it is paired with the presence of mutations from exons 12 and 21. But when acting alone, it does not affect the phenotypic outcome as strongly. An example of exon 26’s haplotype dependency is seen when looking at chemotherapy. Since MDR 1 removes drugs from our cells, inhibitors have been used to block MRD 1's ability to remove drugs, thus letting beneficial drugs like chemotherapy and immunosuppressants aid the body in recovery more efficiently. MDR1 has different proteins that help exile these specific drugs from cancer cells.[26] Verapamil and cyclosporine A are common inhibitors for MDR 1.[23] Unfortunately, when C3435T is mutated with a mutation from either exon 12 or exon 21 (or if all three mutations occur at the same time creating a haplotype), the inhibitors are less likely to weaken the function of MDR1. Multiple silent mutated genes tend to be more resistant against these inhibitors.[26]
Looking at the molecular level, the reason why C3435T in exon 26 of MDR 1 gene is not silent is because of the pace at which the amino acids are being translated to proteins.[25] mRNA’s secondary structures can fold which means different codons correspond to different folding's of the mRNA. For example, when exon 26 changes ATC to ATT both codons produce the same amino acid but ATC is seen more often than the mutation codon. As a consequence, the amount of time it takes for the ribosome to produce its protein confirmation is changed. This leads to a protein structure different from the usual shape of the protein which leads to different functions of the protein.[27]
Other reasons behind MDR1’s “silent mutation” occurs in messenger RNA. In mRNA, codons also work as exon splicing enhancers. Codons decide when to cut out introns based on the codon it is reading in mRNA.[24] The mutated codons have a higher risk of making a mistake when splicing introns out of the mRNA sequence leading to the wrong exons being produced. Therefore, making a change to the mature messenger RNA.[27] Mutations in the Multi-Drug Resistance Gene 1 show how silent mutations can have an effect on the outcome of the phenotype.
See also
References
- Kimchi-Sarfaty C, Oh JM, Kim IW, Sauna ZE, Calcagno AM, Ambudkar SV, Gottesman MM (January 2007). "A "silent" polymorphism in the MDR1 gene changes substrate specificity". Science. 315 (5811): 525–8. doi:10.1126/science.1135308. PMID 17185560. S2CID 15146955.
- Chamary JV, Parmley JL, Hurst LD (February 2006). "Hearing silence: non-neutral evolution at synonymous sites in mammals". Nature Reviews. Genetics. 7 (2): 98–108. doi:10.1038/nrg1770. PMID 16418745. S2CID 25713689.
- Goymer P (February 2007). "Synonymous mutations break their silence". Nature Reviews Genetics. 8 (2): 92. doi:10.1038/nrg2056. S2CID 29882152.
- Zhou T, Ko EA, Gu W, Lim I, Bang H, Ko JH (31 October 2012). "Non-silent story on synonymous sites in voltage-gated ion channel genes". PLOS ONE. 7 (10): e48541. doi:10.1371/journal.pone.0048541. PMC 3485311. PMID 23119053.
- Graur D (2003). "Single Base Mutation" (PDF). In Cooper DN (ed.). Nature Encyclopedia of the Human Genome. MacMillan. ISBN 978-0333803868.
- Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P (2007). Molecular Biology of the Cell. Garland Science. p. 264. ISBN 978-1-136-84442-3.
- Brooker R (2017-02-01). Genetics: Analysis and Principles. McGraw-Hill Higher Education. ISBN 9781259616020.
- "Mutations and Disease | Understanding Genetics". genetics.thetech.org. Retrieved 2018-11-10.
- Watson JD (2008). Molecular Biology of the Gene (6th ed.). San Francisco: Pearson/Benjamin Cummings. ISBN 978-0805395921.
- Angov E (June 2011). "Codon usage: nature's roadmap to expression and folding of proteins". Biotechnology Journal. 6 (6): 650–9. doi:10.1002/biot.201000332. PMC 3166658. PMID 21567958.
- Teng S, Madej T, Panchenko A, Alexov E (March 2009). "Modeling effects of human single nucleotide polymorphisms on protein-protein interactions". Biophysical Journal. 96 (6): 2178–88. doi:10.1016/j.bpj.2008.12.3904. PMC 2717281. PMID 19289044.
- Strachan T, Read AP (1999). Human Molecular Genetics (2nd ed.). Wiley-Liss. ISBN 978-1-85996-202-2. PMID 21089233. NBK7580.
- Czech A, Fedyunin I, Zhang G, Ignatova Z (October 2010). "Silent mutations in sight: co-variations in tRNA abundance as a key to unravel consequences of silent mutations". Molecular BioSystems. 6 (10): 1767–72. doi:10.1039/c004796c. PMID 20617253.
- Komar AA (August 2007). "Silent SNPs: impact on gene function and phenotype". Pharmacogenomics. 8 (8): 1075–80. doi:10.2217/14622416.8.8.1075. PMID 17716239.
- "MIT Biochemistry Lecture Notes-Protein Folding and Human Disease" (PDF).
- "Orders of protein structure". Khan Academy. Retrieved 2018-11-08.
- Shabalina SA, Ogurtsov AY, Spiridonov NA (2006). "A periodic pattern of mRNA secondary structure created by the genetic code". Nucleic Acids Research. 34 (8): 2428–37. doi:10.1093/nar/gkl287. PMC 1458515. PMID 16682450.
- Herschlag D (September 1995). "RNA chaperones and the RNA folding problem". The Journal of Biological Chemistry. 270 (36): 20871–4. CiteSeerX 10.1.1.328.5762. doi:10.1074/jbc.270.36.20871. PMID 7545662. S2CID 14083129.
- Komar AA (January 2007). "Genetics. SNPs, silent but not invisible". Science. 315 (5811): 466–7. doi:10.1126/science.1138239. PMID 17185559. S2CID 41904137.
- Beckman (22 December 2006). "The Sound of a Silent Mutation". News. Science/AAAS.
- Zhang Z, Miteva MA, Wang L, Alexov E (2012). "Analyzing effects of naturally occurring missense mutations". Computational and Mathematical Methods in Medicine. 2012: 1–15. doi:10.1155/2012/805827. PMC 3346971. PMID 22577471.
- Montera M, Piaggio F, Marchese C, Gismondi V, Stella A, Resta N, Varesco L, Guanti G, Mareni C (December 2001). "A silent mutation in exon 14 of the APC gene is associated with exon skipping in a FAP family". Journal of Medical Genetics. 38 (12): 863–7. doi:10.1136/jmg.38.12.863. PMC 1734788. PMID 11768390. Full text
- Weber, Wendell (2008-04-02). Pharmacogenetics. Oxford University Press, USA. ISBN 9780195341515.
- Dudek, Ronald W. (2007). High-yield Cell and Molecular Biology. Lippincott Williams & Wilkins. ISBN 9780781768870.
- Strachan, Tom; Read, Andrew (2018-03-29). Human Molecular Genetics. Garland Science. ISBN 9781136844072.
- "The Sound of a Silent Mutation". Science | AAAS. 2006-12-22. Retrieved 2018-11-18.
- Campbell, Mary K.; Farrell, Shawn O. (2011-01-01). Biochemistry. Cengage Learning. ISBN 978-0840068583.
Further reading
- Mueller S, Coleman JR, Wimmer E (March 2009). "Putting synthesis into biology: a viral view of genetic engineering through de novo gene and genome synthesis". Chemistry & Biology. 16 (3): 337–47. doi:10.1016/j.chembiol.2009.03.002. PMC 2728443. PMID 19318214.
By large-scale computer-aided redesign of the viral genome we engineered hundreds of silent mutations into poliovirus. ... We termed this process of perturbing intrinsic viral genome biases by synthetic genome re-design SAVE for Synthetic Attenuated Virus Engineering
External links
| Wikimedia Commons has media related to Silent mutation. |
- Overview article — Chamary J, Hurst LD (June 2009). "How Trivial DNA Changes Can Hurt Health". Scientific American.
- "WatCut: An on-line tool for restriction analysis, silent mutation scanning, and SNP-RFLP analysis". University of Waterloo. April 17, 2014.
