Therapeutic gene modulation
Therapeutic gene modulation refers to the practice of altering the expression of a gene at one of various stages, with a view to alleviate some form of ailment. It differs from gene therapy in that gene modulation seeks to alter the expression of an endogenous gene (perhaps through the introduction of a gene encoding a novel modulatory protein) whereas gene therapy concerns the introduction of a gene whose product aids the recipient directly.
Modulation of gene expression can be mediated at the level of transcription by DNA-binding agents (which may be artificial transcription factors), small molecules, or synthetic oligonucleotides. It may also be mediated post-transcriptionally through RNA interference.
Transcriptional gene modulation
An approach to therapeutic modulation utilizes agents that modulate endogenous transcription by specifically targeting those genes at the gDNA level. The advantage to this approach over modulation at the mRNA or protein level is that every cell contains only a single gDNA copy. Thus the target copy number is significantly lower allowing the drugs to theoretically be administered at much lower doses.
This approach also offers several advantages over traditional gene therapy. Directly targeting endogenous transcription should yield correct relative expression of splice variants. In contrast, traditional gene therapy typically introduces a gene which can express only one transcript, rather than a set of stoichiometrically-expressed spliced transcript variants. Additionally, virally-introduced genes can be targeted for gene silencing by methylation which can counteract the effect of traditional gene therapy.[1] This is not anticipated to be a problem for transcriptional modulation as it acts on endogenous DNA.
There are three major categories of agents that act as transcriptional gene modulators: triplex-forming oligonucleotides (TFOs), synthetic polyamides (SPAs), and DNA binding proteins.[2]
What are they
Triplex-forming oligonucleotides (TFO) are one potential method to achieve therapeutic gene modulation. TFOs are approximately 10-40 base pairs long and can bind in the major groove in duplex DNA which creates a third strand or a triple helix.[2][3] The binding occurs at polypurine or polypyrimidine regions via Hoogsteen hydrogen bonds to the purine (A / G) bases on the double stranded DNA that is already in the form of the Watson-Crick helix.[4]
How they work
TFOs can be either polypurine or polypyrimidine molecules and bind to one of the two strands in the double helix in either parallel or antiparallel orientation to target polypurine or polypyrimidine regions. Since the DNA-recognition codes are different for the parallel and the anti-parallel fashion of TFO binding, TFOs composed of pyrimidines (C / T) bind to the purine-rich strand of the target double helix via Hoogsteen hydrogen bonds in a parallel fashion.[3] TFOs composed of purines (A / G), or mixed purine and pyrimidine bind to the same purine-rich strand via reverse Hoogsteen bonds in an anti-parallel fashion. TFO's can recognize purine-rich target strands for duplex DNA.[2]

Complications and limitations
In order for TFO motifs to bind in a parallel fashion and create hydrogen bonds, the nitrogen atom at position 3 on the cytosine residue needs to be protonated, but at physiological pH levels it is not, which could prevent parallel binding.[2]
Another limitation is that TFOs can only bind to purine-rich target strands and this would limit the choice of endogenous gene target sites to polypurine-polypyrimidine stretches in duplex DNA. If a method to also allow TFOs to bind to pyrimidine bases was generated, this would enable TFOs to target any part of the genome. Also the human genome is rich in polypurine and polypyrimidine sequences which could affect the specificity of TFO to bind to a target DNA region. An approach to overcome this limitation is to develop TFOs with modified nucleotides that act as locked nucleic acids to increase the affinity of the TFO for specific target sequences.[5]
Other limitations include concerns regarding binding affinity and specificity, in vivo stability, and uptake into cells. Researchers are attempting to overcome these limitations by improving TFO characteristics through chemical modifications, such as modifying the TFO backbone to reduce electrostatic repulsions between the TFO and the DNA duplex. Also due to their high molecular weight, uptake into cells is limited and some strategies to overcome this include DNA condensing agents, coupling of the TFO to hydrophobic residues like cholesterol, or cell permeabilization agents.[2]
What can they do
Scientists are still refining the technology to turn TFOs into a therapeutic product and much of this revolves around their potential applications in antigene therapy. In particular they have been used as inducers of site-specific mutations, reagents that selectively and specifically cleave target DNA, and as modulators of gene expression.[6] One such gene sequence modification method is through the targeting DNA with TFOs to active a target gene. If a target sequence is located between two inactive copies of a gene, DNA ligands, such as TFOs, can bind to the target site and would be recognized as DNA lesions. To fix these lesions, DNA repair complexes are assembled on the targeted sequence, the DNA is repaired. Damage of the intramolecular recombination substrate can then be repaired and detected if resection goes far enough to produce compatible ends on both sides of the cleavage site and then 3' overhangs are ligated leading to the formation of a single active copy of the gene and the loss of all the sequences between the two copies of the gene.[4]
In model systems TFOs can inhibit gene expression at the DNA level as well as induce targeted mutagenesis in the model.[6] TFO-induced inhibition of transcription elongation on endogenous targets have been tested on cell cultures with success.[7] However, despite much in vitro success, there has been limited achievement in cellular applications potentially due to target accessibility.
TFOs have the potential to silence silence gene by targeting transcription initiation or elongation, arresting at the triplex binding sites, or introducing permanent changes in a target sequence via stimulating a cell's inherent repair pathways. These applications can be relevant in creating cancer therapies that inhibit gene expression at the DNA level. Since aberrant gene expression is a hallmark of cancer, modulating these endogenous genes' expression levels could potentially act as a therapy for multiple cancer types.
Synthetic polyamides
Synthetic polyamides are a set of small molecules that form specific hydrogen bonds to the minor groove of DNA. They can exert an effect either directly, by binding a regulatory region or transcribed region of a gene to modify transcription, or indirectly, by designed conjugation with another agent that makes alterations around the DNA target site.
Structure
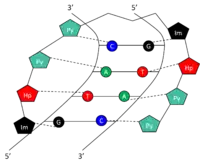
Specific bases in the minor groove of DNA can be recognized and bound by small synthetic polyamides (SPAs). DNA-binding SPAs have been engineered to contain three polyamide amino acid components: hydroxypyrrole (Hp), imidazole (Im), and pyrrole (Py).[10] Chains of these amino acids loop back on themselves in a hairpin structure. The amino acids on either side of the hairpin form a pair which can specifically recognize both sides of a Watson-Crick base pair. This occurs through hydrogen bonding within the minor groove of DNA. The amide pairs Py/Im, Py/Hp, Hp/Py, and Im/Py recognize the Watson-Crick base pairs C-G, A-T, T-A, and G-C, respectively (Table 1). See figure for a graphical representation of 5'-GTAC-3' recognition by a SPA. SPAs have low toxicity, but have not yet been used in human gene modulation.
| Amide Pair | Nucleotide Pair |
|---|---|
| Py/Im | C-G |
| Py/Hp | A-T |
| Hp/Py | T-A |
| Im/Py | G-C |
Limitations and workarounds
The major structural drawback to unmodified SPAs as gene modulators is that their recognition sequence cannot be extended beyond 5 Watson-Crick base pairings. The natural curvature of the DNA minor groove is too tight a turn for the hairpin structure to match. There are several groups with proposed workarounds to this problem.[8][11][12][13][14] SPAs can be made to better follow the curvature of the minor groove by inserting beta-alanine which relaxes the structure.[10] Another approach to extending the recognition length is to use several short hairpins in succession.[15][16] This approach has increased the recognition length to up to eleven Watson-Crick base pairs.


Direct modulation
SPAs may inhibit transcription through binding within a transcribed region of a target gene. This inhibition occurs through blocking of elongation by an RNA polymerase.
SPAs may also modulate transcription by targeting a transcription regulator binding site. If the regulator is an activator of transcription, this will decrease transcriptional levels. As an example, SPA targeting to the binding site for the activating transcription factor TFIIIA has been demonstrated to inhibit transcription of the downstream 5S RNA.[17] In contrast, if the regulator is a repressor, this will increase transcriptional levels. As an example, SPA targeting to the host factor LSF, which represses expression of the human immunodeficiency virus (HIV) type 1 long terminal repeat (LTR), blocks binding of LSF and consequently de-represses expression of LTR[18] .

Conjugate modulation
SPAs have not been shown to directly modify DNA or have activity other than direct blocking of other factors or processes. However, modifying agents can be bound to the tail ends of the hairpin structure. The specific binding of the SPA to DNA allows for site-specific targeting of the conjugated modifying agent.
SPAs have been paired with the DNA-alkylating moieties cyclopropylpyrroloindole[19] and chlorambucil[20] that were able to damage and crosslink SV40 DNA. This effect inhibited cell cycling and growth. Chlorambucil, a chemotherapeutic agent, was more effective when conjugated to an SPA than without.
In 2012, SPAs were conjugated to SAHA, a potent histone deacetylase (HDAC) inhibitor.[21] SPAs with conjugated SAHA were targeted to Oct-3/4 and Nanog which induced epigenetic remodeling and consequently increased expression of multiple pluripotency related genes in mouse embryonic fibroblasts.
What they are/structure
Designer zinc-finger proteins are engineered proteins used to target specific areas of DNA. These proteins capitalize on the DNA-binding capacity of natural zinc-finger domains to modulate specific target areas of the genome.[22] In both designer and natural zinc-finger motifs, the protein consists of two β-sheets and one α-helix. Two histidine residues on the α-helix and two cysteine residues on the β-sheets are bonded to a zinc atom, which serves to stabilize the protein domain as a whole. This stabilization particularly benefits the α-helix in its function as the DNA-recognition and -binding domain. Transcription factor TFIIIA is an example of a naturally-occurring protein with zinc-finger motifs.[23]
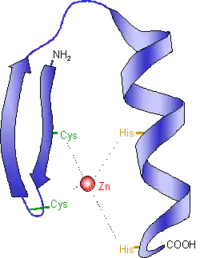
How they work
Zinc-finger motifs bind into the major groove of helical DNA,[23] where the amino acid residue sequence on the α-helix gives the motif its target sequence specificity. The domain binds to a seven-nucleotide sequence of DNA (positions 1 through 6 on the primary strand of DNA, plus positions 0 and 3 on the complementary strand), thereby ensuring that the protein motif is highly selective of its target.[22] In engineering a designer zinc-finger protein, researchers can utilize techniques such as site-directed mutagenesis followed by randomized trials for binding capacity,[22][24] or the in vitro recombination of motifs with known target specificity to produce a library of sequence-specific final proteins.[25]
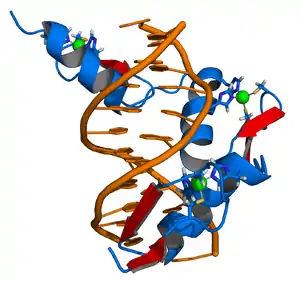
Effects and impacts on gene modulation
Designer zinc-finger proteins can modulate genome expression in a number of ways. Ultimately, two factors are primarily responsible for the end result on expression: whether the targeted sequence is a regulatory region or a coding region of DNA, and whether and what types of effector domains are bound to the zinc-finger domain. If the target sequence for an engineered designer protein is a regulatory domain - e.g., a promoter or a repressor of replication - the binding site for naturally-occurring transcription factors will be obscured, leading to a corresponding decrease or increase, respectively, in transcription for the associated gene.[26] Similarly, if the target sequence is an exon, the designer zinc-finger will obscure the sequence from RNA polymerase transcription complexes, resulting in a truncated or otherwise nonfunctional gene product.[22]
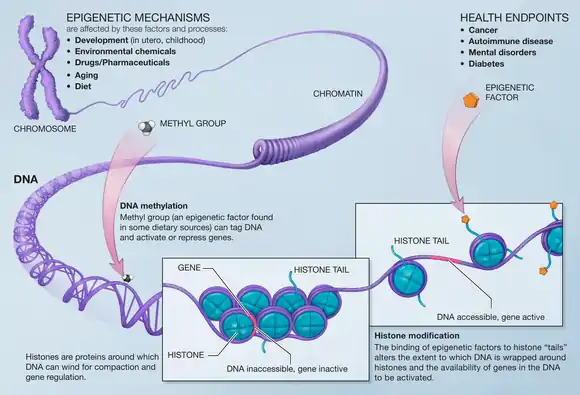
Effector domains bound to the zinc-finger can also have comparable effects. It is the function of these effector domains which are arguably the most important with respect to the use of designer zinc-finger proteins for therapeutic gene modulation. If a methylase domain is bound to the designer zinc-finger protein, when the zinc-finger protein binds to the target DNA sequence an increase in methylation state of DNA in that region will subsequently result. Transcription rates of genes so-affected will be reduced.[27] Many of the effector domains function to modulate either the DNA directly - e.g. via methylation, cleaving,[28] or recombination of the target DNA sequence[29] - or by modulating its transcription rate - e.g. inhibiting transcription via repressor domains that block transcriptional machinery,[30] promoting transcription with activation domains that recruit transcriptional machinery to the site,[31] or histone- or other epigenetic-modification domains that affect chromatin state and the ability of transcriptional machinery to access the affected genes.[32] Epigenetic modification is a major theme in determining varying expression levels for genes, as explained by the idea that how tightly-wound the DNA strand is - from histones at the local level up to chromatin at the chromosomal level - can influence the accessibility of sequences of DNA to transcription machinery, thereby influencing the rate at which it can be transcribed.[23] If, instead of impacting the DNA strand directly, as described above, a designer zinc-finger protein instead affects epigenetic modification state for a target DNA region, modulation of gene expression could similarly be accomplished.
In the first case to successfully demonstrate the use of designer zinc-finger proteins to modulate gene expression in vivo, Choo et al[26] designed a protein consisting of three zinc-finger domains that targeted a specific sequence on a BCR-ABL fusion oncogene. This specific oncogene is implicated in acute lymphoblastic leukemia. The oncogene typically enables leukemia cells to proliferate in the absence of specific growth factors, a hallmark of cancer. By including a nuclear localization signal with the tri-domain zinc-finger protein in order to facilitate binding of the protein to genomic DNA in the nucleus, Choo et al were able to demonstrate that their engineered protein could block transcription of the oncogene in vivo. Leukemia cells became dependent on regular growth factors, bringing the cell cycle back under the control of normal regulation.[26]
Post-transcriptional gene modulation
The major approach to post-transcriptional gene modulation is via RNA interference (RNAi). The primary problem with using RNAi in gene modulation is drug delivery to target cells.[33][34] RNAi gene modulation has been successfully applied to mice toward the treatment of a mouse model for inflammatory bowel disease.[35] This treatment utilized liposome-based beta-7 integrin-targeted, stabilized nanoparticles entrapping short interfering RNAs (siRNAs). There are several other forms of RNAi delivery, including: polyplex delivery, ligand-siRNA conjugates, naked delivery, inorganic particle deliver using gold nanoparticles, and site specific local delivery.[36]
Clinical significance
Designer zinc-finger proteins, on the other hand, have undergone some trials in the clinical arena. The efficacy and safety of EW-A-401, an engineered zinc-finger transcription factor, as a pharmacologic agent for treating claudication, a cardiovascular ailment, has been investigated in clinical trials.[37] The protein consists of an engineered plasmid DNA that prompts the patient to produce an engineered transcription factor, the target of which is the vascular endothelial growth factor-A (VEGF-A) gene, which positively influences blood vessel development. Although not yet approved by the U.S. Food and Drug Administration (FDA), two Phase I clinical studies have been completed which identify this zinc-finger protein as a promising and safe potential therapeutic agent for treatment of peripheral arterial disease in humans.[38]
References
- Young WB, Link CJ (2000). "Chimeric retroviral helper virus and picornavirus IRES sequence to eliminate DNA methylation for improved retroviral packaging cells". Journal of Virology. 74 (11): 5242–5249. doi:10.1128/JVI.74.11.5242-5249.2000. PMC 110878. PMID 10799600.
- Uil TG, Haisma HJ, Rots MG (2003). "Therapeutic modulation of endogenous gene function by agents with designed DNA-sequence specificities". Nucleic Acids Research. 31 (21): 6064–6078. doi:10.1093/nar/gkg815. PMC 275457. PMID 14576293.
- Sargent, RG.; Kim, S.; Gruenert, DC. (2011). "Oligo/polynucleotide-based gene modification: strategies and therapeutic potential". Oligonucleotides. 21 (2): 55–75. doi:10.1089/oli.2010.0273. PMC 3078494. PMID 21417933.
- Simon, P.; Cannata, F.; Concordet, JP.; Giovannangeli, C. (August 2008). "Targeting DNA with triplex-forming oligonucleotides to modify gene sequence". Biochimie. 90 (8): 1109–16. doi:10.1016/j.biochi.2008.04.004. PMID 18460344.
- Zhou, Y.; Kierzek, E.; Loo, ZP.; Antonio, M.; Yau, YH.; Chuah, YW.; Geifman-Shochat, S.; Kierzek, R.; Chen, G. (July 2013). "Recognition of RNA duplexes by chemically modified triplex-forming oligonucleotides". Nucleic Acids Res. 41 (13): 6664–73. doi:10.1093/nar/gkt352. PMC 3711454. PMID 23658228.
- Guntaka, RV.; Varma, BR.; Weber, KT. (January 2003). "Triplex-forming oligonucleotides as modulators of gene expression". Int J Biochem Cell Biol. 35 (1): 22–31. doi:10.1016/s1357-2725(02)00165-6. PMID 12467644.
- Faria, M.; Wood, CD.; Perrouault, L.; Nelson, JS.; Winter, A.; White, MR.; Helene, C.; Giovannangeli, C. (April 2000). "Targeted inhibition of transcription elongation in cells mediated by triplex-forming oligonucleotides". Proc Natl Acad Sci U S A. 97 (8): 3862–7. doi:10.1073/pnas.97.8.3862. PMC 18107. PMID 10760257.
- Reddy BS, Sharma SK, Lown JW (2001). "Recent developments in sequence selective minor groove DNA effectors". Curr. Med. Chem. 8 (5): 475–508. doi:10.2174/0929867003373292. PMID 11281837.
- Dervan PB (2001). "Molecular Recognition of DNA by Small Molecules". Bioorg. Med. Chem. 9 (9): 2215–2235. doi:10.1016/s0968-0896(01)00262-0. PMID 11553460.
- White S, Szewcxyk JW, Turner JM, Baird EE, Dervan PB (1998). "Recognition of the four Watson-Crick base pairs in the DNA minor groove by synthetic ligands" (PDF). Nature. 391 (6666): 468–471. doi:10.1038/35106. PMID 9461213. S2CID 205023593.
- Dervan PB, Edelson BS (2003). "Recognition of the DNA minor groove by pyrrole-imidazole polyamides". Curr. Opin. Struct. Biol. 13 (3): 284–299. doi:10.1016/s0959-440x(03)00081-2. PMID 12831879.
- Lown JW (1988). "Lexitropsins: rational design of DNA sequence reading agents as novel anti-cancer agents and potential cellular probes". Anticancer Drug Des. 3 (1): 25–40. PMID 2838035.
- Trauger JW, Baird EE, Dervan PB (1996). "Recognition of DNA by designed ligands at subnanomolar concentrations". Nature. 382 (6591): 559–561. doi:10.1038/382559a0. PMID 8700233. S2CID 4335955.
- Wemmer DE (2000). "Designed sequence-specific minor groove ligands". Annu. Rev. Biophys. Biomol. Struct. 29: 439–461. doi:10.1146/annurev.biophys.29.1.439. PMID 10940255.
- Kers I, Dervan PB (2002). "Search for the optimal linker in tandem hairpin polyamides". Bioorg. Med. Chem. 10 (10): 3339–3349. doi:10.1016/s0968-0896(02)00221-3. PMID 12150881.
- Weyermann P, Dervan PB (2002). "Recognition of ten base pairs of DNA by head-to-head hairpin dimers" (PDF). J. Am. Chem. Soc. 124 (24): 6872–6878. doi:10.1021/ja020258k. PMID 12059208.
- Gottesfeld JM, Neely L, Trauger JW, Baird EE, Dervan PB (1997). "Regulation of gene expression by small molecules". Nature. 387 (6629): 202–205. doi:10.1038/387202a0. PMID 9144294. S2CID 4358491.
- Coull JJ, He G, Melander C, Rucker VC, Dervan PB, Margolis DM (2002). "Targeted derepression of the human immunodeficiency virus type 1 long terminal repeat by pyrrole-imidazole polyamides". J. Virol. 76 (23): 12349–12354. doi:10.1128/jvi.76.23.12349-12354.2002. PMC 136904. PMID 12414976.
- Wang YD, Dziegielewski J, Wurtz NR, Dziegielewska B, Dervan PB, Beerman TA (2003). "DNA crosslinking and biological activity of a hairpin polyamide-chlorambucil conjugate". Nucleic Acids Research. 31 (21): 1208–1215. doi:10.1093/nar/gkg215. PMC 150233. PMID 12582240.
- Wang YD, Dziegielewski J, Chang AY, Dervan PB, Beerman TA (2002). "Cell-free and cellular activities of a DNA sequence selective hairpin polyamide-CBI conjugate". J. Biol. Chem. 277 (45): 42431–42437. doi:10.1074/jbc.M207179200. PMID 12196541.
- Pandian NG, Nakano Y, Sato S, Morinaga H, Bando T, Nagase H, Sugiyama H (2012). "A synthetic small molecule for rapid induction of multiple pluripotency genes in mouse embryonic fibroblasts". Scientific Reports. 2 (544): 544. doi:10.1038/srep00544. PMC 3408130. PMID 22848790.
- Papworth, M.; Kolasinska, P.; Minczuk, M. (January 2006). "Designer zinc-finger proteins and their applications". Gene. 366 (1): 27–38. doi:10.1016/j.gene.2005.09.011. PMID 16298089.
- Watson, James D. (2008). Molecular biology of the gene. San Francisco: Pearson/Benjamin Cummings. p. 595. ISBN 978-0-8053-9592-1.
- Desjarlais, JR.; Berg, JM. (March 1993). "Use of a zinc-finger consensus sequence framework and specificity rules to design specific DNA binding proteins". Proc Natl Acad Sci U S A. 90 (6): 2256–60. doi:10.1073/pnas.90.6.2256. PMC 46065. PMID 8460130.
- Isalan, M.; Klug, A.; Choo, Y. (July 2001). "A rapid, generally applicable method to engineer zinc fingers illustrated by targeting the HIV-1 promoter". Nat Biotechnol. 19 (7): 656–60. doi:10.1038/90264. PMC 2677679. PMID 11433278.
- Choo, Y.; Sánchez-García, I.; Klug, A. (December 1994). "In vivo repression by a site-specific DNA-binding protein designed against an oncogenic sequence" (PDF). Nature. 372 (6507): 642–5. doi:10.1038/372642a0. hdl:10261/6295. PMID 7990954. S2CID 12701336.
- Carvin, CD.; Parr, RD.; Kladde, MP. (November 2003). "Site-selective in vivo targeting of cytosine-5 DNA methylation by zinc-finger proteins". Nucleic Acids Res. 31 (22): 6493–501. doi:10.1093/nar/gkg853. PMC 275549. PMID 14602907.
- Kim, YG.; Cha, J.; Chandrasegaran, S. (February 1996). "Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain". Proc Natl Acad Sci U S A. 93 (3): 1156–60. doi:10.1073/pnas.93.3.1156. PMC 40048. PMID 8577732.
- Urnov, FD.; Miller, JC.; Lee, YL.; Beausejour, CM.; Rock, JM.; Augustus, S.; Jamieson, AC.; Porteus, MH.; et al. (June 2005). "Highly efficient endogenous human gene correction using designed zinc-finger nucleases". Nature. 435 (7042): 646–51. doi:10.1038/nature03556. PMID 15806097. S2CID 4390010.
- Beerli, RR.; Dreier, B.; Barbas, CF. (February 2000). "Positive and negative regulation of endogenous genes by designed transcription factors". Proc Natl Acad Sci U S A. 97 (4): 1495–500. doi:10.1073/pnas.040552697. PMC 26462. PMID 10660690.
- Lara, H.; Wang, Y.; Beltran, AS.; Juárez-Moreno, K.; Yuan, X.; Kato, S.; Leisewitz, AV.; Cuello Fredes, M.; et al. (August 2012). "Targeting serous epithelial ovarian cancer with designer zinc finger transcription factors". J Biol Chem. 287 (35): 29873–86. doi:10.1074/jbc.M112.360768. PMC 3436144. PMID 22782891.
- Snowden, AW.; Zhang, L.; Urnov, F.; Dent, C.; Jouvenot, Y.; Zhong, X.; Rebar, EJ.; Jamieson, AC.; et al. (December 2003). "Repression of vascular endothelial growth factor A in glioblastoma cells using engineered zinc finger transcription factors". Cancer Res. 63 (24): 8968–76. PMID 14695215.
- Behlke, MA. (April 2006). "Progress towards in vivo use of siRNAs". Mol Ther. 13 (4): 644–70. doi:10.1016/j.ymthe.2006.01.001. PMC 7106286. PMID 16481219.
- Dykxhoorn, DM.; Lieberman, J. (2006). "Running interference: prospects and obstacles to using small interfering RNAs as small molecule drugs". Annu Rev Biomed Eng. 8: 377–402. CiteSeerX 10.1.1.418.758. doi:10.1146/annurev.bioeng.8.061505.095848. PMID 16834561.
- Peer, D.; Park, EJ.; Morishita, Y.; Carman, CV.; Shimaoka, M. (February 2008). "Systemic leukocyte-directed siRNA delivery revealing cyclin D1 as an anti-inflammatory target". Science. 319 (5863): 627–30. doi:10.1126/science.1149859. PMC 2490797. PMID 18239128.
- Rettig, GR.; Behlke, MA. (March 2012). "Progress toward in vivo use of siRNAs-II". Mol Ther. 20 (3): 483–512. doi:10.1038/mt.2011.263. PMC 3293614. PMID 22186795.
- "Identifier NCT00080392.Modulation of Vascular Endothelial Growth Factor (VEGF) Using an Engineered Zinc-Finger Transcription Factor to Treat Lower Limb Intermittent Claudication". ClinicalTrials.gov. U.S. National Institutes of Health. 30 December 2011. Retrieved 25 July 2013.
- Giacca, M.; Zacchigna, S. (June 2012). "VEGF gene therapy: therapeutic angiogenesis in the clinic and beyond". Gene Ther. 19 (6): 622–9. doi:10.1038/gt.2012.17. PMID 22378343.