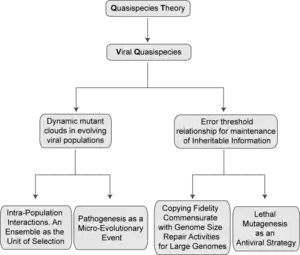
Viral quasispecies
A viral quasispecies is a population structure of viruses with a large numbers of variant genomes (related by mutations). Quasispecies result from high mutation rates as mutants arise continually and change in relative frequency as viral replication and selection proceeds.
The theory predicts that a viral quasispecies at a low but evolutionarily neutral and highly connected (that is, flat) region in the fitness landscape will outcompete a quasispecies located at a higher but narrower fitness peak in which the surrounding mutants are unfit.[1][2] This phenomenon has been called 'the quasispecies effect' or, more recently, the 'survival of the flattest'.[3]
The term quasispecies was adopted from a theory of the origin of life in which primitive replicons consisted of mutant distributions, as found experimentally with present-day RNA viruses within their host.[4][5] The theory provided a new definition of wild type when describing viruses, and a conceptual framework for the interpretation of the adaptive potential of RNA viruses that contrasted with classical studies based on consensus sequences.
The quasispecies model is most applicable when the genome size is limited and the mutation rate is high, and so is most relevant to RNA viruses (including important pathogens) because they have high mutation rates (approx one error per round of replication),[6] though the concepts can apply to other biological entities. In such scenarios, complex distributions of closely related variant genomes are subjected to genetic variation, competition and selection, and may act as a unit of selection. Therefore, the evolutionary trajectory of the viral infection cannot be predicted solely from the characteristics of the fittest sequence. High mutation rates also place an upper limit compatible with inheritable information. Crossing such a limit leads to RNA virus extinction, a transition that is the basis of an antiviral design termed lethal mutagenesis, and of relevance to antiviral medicine.
The relevance of quasispecies in virology has been the subject of extended debate. However, standard clonal analyses and deep sequencing methodologies have confirmed the presence of myriads of mutant genomes in viral populations, and their participation in adaptive processes.
History
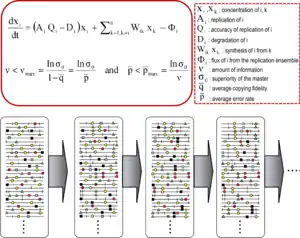
Quasispecies theory was developed in the 1970s by Manfred Eigen and Peter Schuster to explain self-organization and adaptability of primitive replicons (a term used to refer to any replicating entity), as an ingredient of hypercyclic organizations that link genotypic and phenotypic information, as an essential step in the origin of life.[9][7] The theory portrayed early replicon populations as organized mutant spectra dominated by a master sequence, the one endowed with the highest fitness (replicative capacity) in the distribution. It introduced the notion of a mutant ensemble as a unit of selection, thus emphasizing the relevance of intra-population interactions to understand the response to selective constraints. One of its corollaries is the error threshold relationship, which marks the maximum mutation rate at which the master (or dominant) sequence can stabilize the mutant ensemble. Violation of the error threshold results in loss of dominance of the master sequence and drift of the population in sequence space.[7][10][11][12]
The core quasispecies concepts are described by two fundamental equations: replication with production of error copies, and the error threshold relationship. They capture two major features of RNA viruses at the population level: the presence of a mutant spectrum, and the adverse effect of an increase of mutation rate on virus survival, each with several derivations.
The existence of a mutant spectrum was experimentally evidenced first by clonal analyses of RNA bacteriophage Qβ populations whose replication had been initiated by a single virus particle. Individual genomes differed from the consensus sequence in an average of one to two mutations per individual genome.[13] Fitness of biological clones was inferior to that of the parental, uncloned population, a difference also documented for vesicular stomatitis virus (VSV).[14] The replicative capacity of a population ensemble need not coincide with that of its individual components. The finding that a viral population was essentially a pool of mutants came at a time when mutations in general genetics were considered rare events, and virologists associated a viral genome with a defined nucleotide sequence, as still implied today in the contents of data banks.[15] The cloud nature of Qβ was understood as a consequence of its high mutation rate, calculated in 10−4 mutations introduced per nucleotide copied,[16] together with tolerance of individual genomes to accept an undetermined proportion of the newly arising mutations, despite fitness costs. The error rate estimated for bacteriophage Qβ has been confirmed, and is comparable to values calculated for other RNA viruses.[6][17]
High mutation rates and quasispecies were verified for other RNA viruses based on dissection of viral populations by molecular or biological cloning, and sequence analysis of individual clones. John Holland and colleagues were the first to recognize that a rapidly evolving RNA world inserted in a DNA-based biosphere had multiple evolutionary and medical implications.[14][18][19][20] Genome plasticity of RNA viruses had been suspected for many decades. Key early observations were variations in viral traits described by Findley in the 1930s, the studies of Granoff on transitions of plaque morphology of Newcastle disease virus, or the high frequency of conversions between drug resistance and dependence in Coxsackie A9 virus, among other studies with animal and plant viruses in the middle of the 20th century.[21] When put in the context of present-day knowledge, we realize that these observations on phenotypic changes were the tip of the iceberg of an extremely complex reality of viral populations. High mutation rates and population heterogeneity characterize RNA viruses, with consequences for viral pathogenesis and the control of viral disease. Detailed studies on quasispecies dynamics in vivo have been performed with human immunodeficiency virus type 1 (HIV-1) and hepatitis C virus.[8][22][23]
Current scope
The first mathematical formulation of quasispecies was deterministic; it assumed steady state mutant distributions in genetic equilibrium without perturbations derived from modifications of the environment or population size.[24] These conditions are common in initial theoretical formulations of complex phenomena because they confer mathematical tractability. Since then, several extensions of the theory to non-equilibrium conditions with stochastic components have been developed, with the aim of finding general solutions for multi-peak fitness landscapes. These objectives approximate quasispecies to the real case of RNA viruses, which are compelled to deal with dramatic variations in population size and environment.[25] Research on quasispecies has proceeded through several theoretical and experimental avenues that include continuing studies on evolutionary optimization and the origin of life, RNA-RNA interactions and replicator networks, the error threshold in variable fitness landscapes, consideration of chemical mutagenesis and proofreading mechanisms, evolution of tumor cells, bacterial populations or stem cells, chromosomal instability, drug resistance, and conformation distributions in prions (a class of proteins with conformation-dependent pathogenic potential; in this case the quasispecies is defined by a distribution of conformations).[8][26] New inputs into experimental quasispecies research have come from deep sequencing to probe viral and cellular populations, recognition of interactions within mutant spectra, models of viral population dynamics related to disease progression and pathogen transmission, and new teachings from fidelity variants of viruses.[26] Here we summarize the main aspects of quasispecies dynamics, and recent developments relevant to virus evolution and pathogenesis.
Dynamic heterogeneity
The molecular basis of high error rates is the limited template-copying fidelity of RNA-dependent RNA polymerases (RdRps) and RNA-dependent DNA polymerases (also termed reverse transcriptases, RTs). In addition, these enzymes are defective in proofreading[27] because they lack a 3’ to 5’ exonuclease domain present in replicative cellular DNA polymerases.[28] Also, postreplicative-repair pathways, abundant to correct genetic lesions in replicating cellular DNA, appear as ineffective for double-stranded RNA or RNA-DNA hybrids. The presence of a proofreading-repair activity in coronaviruses increases their copying accuracy in about 15-fold.[29] This and other repair activities, that may act on standard RNA or retroviral genomes,[30][31][32][33] do not prevent the formation of mutant spectra, although their amplitude may be lower than for other RNA viruses, at least in populations close to a clonal (single genome) origin. Quasispecies dynamics will operate in any viral or cellular system in which due to high mutation rates (as a result of low fidelity nucleic acid polymerases or environmental alterations) mutant spectra are rapidly generated.[8][34][35][36][37][38]
Studies with different virus-host systems have established some general observations on the mechanisms of mutant generation, and implications of quasispecies dynamics.[8][39][40][41][42][43][44][45][46][47][48] In RNA virus genetics when we speak of “a mutant” the entity we handle is a cloud of mutants in which the specific mutation to which we direct our attention is present in all (or the great majority of) individual genomes. There is no such a thing as “a” wild type or “a” mutant virus. They are always clouds of mutants. Changes in the relative dominance of components of mutant spectra are particularly severe during in vivo infections, with complex dynamics of intra-host heterogeneity and variations. Bioinformatic procedures have been developed to unveil the relationships among different but closely related genome types that may suggest some hierarchical order of mutation acquisition or identification of transmission clusters (examples are Partition Analysis of Quasispecies, PAQ[49] or QUasispecies Evolution, Network-based Transmission Inference, QUENTIN[50]).
Phenotypic reservoirs
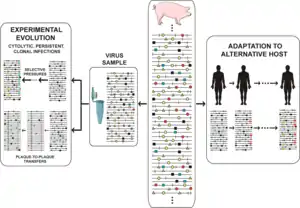
The crux of the matter regarding quasispecies implications is that at any given time, the viral population includes a reservoir not only of genotypic but also of phenotypic variants, conferring upon the population some adaptive pluripotency. Accumulating laboratory and clinical evidence renders untenable that minority components of mutant spectra should be dismissed on the grounds of their being neutral. They can participate in selective processes and cannot be excluded from interpretations of virus behavior. Variation universally involves point mutations and it can also include recombination (in its replicative and non-replicative modes), and genome segment reassortment.[40] All modes of molecular variation are compatible, only restricted by the scope of mechanisms accessible to the replicative machinery, and for the need for viral genomes to remain functional. David Evans and colleagues identified many recombination events associated with enterovirus replication, and only a few recombinants made their way towards continued replication.[51] Recombination can mediate adaptability and virulence.[52] High mutation and recombination rates have led to the conceptual distinction between mechanistically unavoidable and evolutionarily relevant variation, in connection with the issue of clonal versus non-clonal nature of virus evolution (microbial evolution in general).[53][54] Only a minority of the nascent variation during replication can be successfully propagated. Within limits that are set by biological constraints, each population is made of an array of variant genomes, with a total number which is commensurate with the virus population size. To infect a plant, animal or cell culture with 103 infectious units can have very different consequences than to infect with 1010 infectious units, not only because the host defense systems may be overwhelmed by the high infectious dose, but also because the mutant repertoire that engages in adaptive explorations is larger. Part of the variants of a mutant spectrum, either in isolation or in consortium with others,[55] may perform better than other members of the same population in the event of an environmental change. Selective pressures favor replication of some components of a mutant spectrum over others, despite all of them being interconnected by mutation. Differential performance can be at the level of viral genomes (during replication, intracellular gene expression, interaction with host factors, etc.) or viral particles (for thermal stability, entry into or exit from cells, to withstand neutralizing antibodies, etc.).[20][8][21][22][23][40][41][42] Adaptability of RNA viruses is linked to parameters that facilitate exploration of sequence space: genome size (1.8 to 33 Kb), population size (variable but that can attain an impressive 1012 individual genomes in an infected host at a given time), replication rate, mutation rate, fecundity (yield of viral particles per cell), and number of mutations required for a phenotypic change (surprisingly low for several relevant traits[56]).
Mutant spectrum dynamics has been depicted in different ways, and we have chosen one that encompasses frequent events in natural populations and research designs, such as virus isolation from an infected host, adaptation to cell culture for studies on experimental evolution, or adaptation to alternative hosts in vivo. The reality is even more complex, given the large population sizes, with an indeterminate proportion of genomes actively replicating at any given time (sometimes equated with the effective population size in general genetics), and harboring multiple mutations per genome. The scenarios suggested by current experimental data defy our imagination. The relative frequency of individual mutations fluctuates in an unceasing exploration of sequence space,[57][58][8] with phenotypic changes (not only genotypic changes) being far more frequent than previously thought. The experimental evolution design that consists of passaging viral populations for long time periods (many sequential infections) is often extremely revealing. In foot-and-mouth disease virus (FMDV) such a design led to a remarkable phenotypic diversification into subpopulations of colonizers and competitors, that modulated virulence of the mutant ensemble.[59] In HCV such a design unveiled continuous mutation waves and a more accurate understanding of the types of fitness landscapes occupied by high fitness viruses.[58][60]
Limitations and indeterminacies
The nucleotide sequence of an individual genome from a population (no matter which the degree of population complexity might be), can be determined either following a biological or molecular cloning event or by deep sequencing of entire viral genomes, in a manner that mutation linkage (assignment of different mutations to the same genome molecule) can be established. Each of these procedures implies some limitations: biological cloning can bias the representation in favor of infectious genomes, while molecular cloning can introduce non-infectious (defective) genomes in the analysis.[20][56][57] Whole genome quasispecies description is still technically challenging due to the artifactual introduction of mutations. Most current deep sequencing platforms yield sequences of short reads for a given amplicon (sequence under analysis); minority mutations in an amplicon cannot be reliably linked to mutations in a different amplicon of the same genome; at most, statistical inferences on linkage can be proposed. Despite these limitations, control experiments and improvements of bioinformatic procedures support that the majority of sequence heterogeneity analyzed in viral populations indeed reflects differences in the natural template populations. If mutation linkage can be solved on a routine basis, a new wave of molecular information relevant to epistatic interactions will enter the picture.
There are additional levels of indeterminacy in the sequential analysis of viral populations, in particular those replicating in vivo. Components of the mutant spectrum represented at a given time in the sample taken for sequencing may differ from those in the next time point, due either to sampling uncertainties or bona fide fluctuations of genome frequencies. It is not justified to accept a rough similarity because even a single mutation in a given sequence context may affect biological properties.[8] In the words of John Holland and colleagues: “It is important to remember that every quasispecies genome swarm in an infected individual is unique and “new” in the sense that no identical population of genomes has ever existed before and none such will ever exist again”.[61] On top of the fleeting nature of any mutant distribution, the standard methods available for quasispecies characterization provide genomic sequences of a minority of the population (estimated in 10−8 to 10−13 for molecular cloning-Sanger sequencing, and in 10−6 to 10−11 for deep sequencing).[56] We can only have an approximate representation of viral populations and their dynamics, as evidenced by many experimental studies.[13][18][8][41][42][56][58][62]
Non-consensus-based descriptors
The points summarized in previous sections fully justifies addressing analytical tools towards the mutant spectrum rather than ignoring it or considering its presence a side issue. Use of consensus sequences to describe the genome of a virus isolate, despite being warranted by the difficulties of conveying the information recapitulated in a mutant spectrum, blurs and enfeebles biological interpretations. Experimental results have demonstrated that minority genomes from a mutant spectrum (that cannot be identified by examining the consensus sequence) can include mutations that confer resistance to antiviral inhibitors, neutralizing antibodies or cytotoxic T cells, or that can alter the capacity to induce interferon (IFN) or to respond to IFN, virulence or particle stability, among other phenotypic traits.[8][40][44][58][62][63][64][65] Mutant spectra can also mediate cyclical adaptation to different cell types.[39] A mutant spectrum defines a consensus but the consensus is an abstraction; it may not be represented in the population. Many events in viral pathogenesis and evolution are due to mutant spectrum modifications or interactions which cannot be properly interpreted solely on the basis of consensus sequences.[13][8][20][21][26][39][38][41][42][51][52][55][58][61]
Collective response
Mutant spectra are not mere aggregates of mutants acting independently. They are often engaged in collective responses of two major types: those that depend on the presence of sets of variants, and those that rely on intra-mutant spectrum interactions.
Behavior of reconstructed quasispecies
In some cases of sweeping selection (very strong selection for a trait), an individual (or a limited number of individuals) that encodes signatures prone to be selected, may approach dominance while becoming the founder of a mutant cloud (because formation of a cloud is inherent to replication). Conditions for dominance (in this case in response to selection) are that the genome senses the selective sweep and that its replication in the new selective environment is permitted. In other cases, a collection of mutants is selected. This was illustrated with a FMDV quasispecies that was reconstructed in the laboratory with multiple antigenic variants (each at low frequency) that belonged to two different categories, and shared resistance to the same monoclonal antibody.[66] One category included mutants with an amino acid substitution that affected receptor recognition (since the antigenic determinant overlapped with the integrin receptor recognition site); in the other category, the substitutions affected the antigenic determinant but not the receptor recognition site. Passages of the virus in absence of the monoclonal antibody resulted in dominance of antigenic variants that maintained the receptor recognition capacity, but the dominant variants were surrounded by a cloud of mutants of the other antigenic variant category. Conversely, passages in the presence of the antibody led to selection of variants with altered receptor recognition, surrounded by a cloud of antigenic variants that maintained receptor recognition. The results underlined the role of mutant clouds in selective events, and unveiled a new mechanism of antigenic flexibility.[66]
Quasispecies memory
Quasispecies memory is a type of molecular memory dependent on the recent history of the evolutionary lineage and the integrity of the mutant spectrum.[67][68] The search for memory was prompted by the complex adaptive system behavior of a viral quasispecies, suggested by the presence of core information (considered the one that defines viral identity) despite variation of constitutive elements (the mutant spectrum). A well-known example is memory in the immune system that mobilizes and expands minority components in response to stimuli previously faced by the system.[69] In the experiments designed to identify memory in viral quasispecies, members of the mutant spectrum increased in frequency as a consequence of their replication during a selection event that drove them towards dominance. When the selective constraint was withdrawn, memory genomes remained at levels that were 10- to 100-fold higher than the basal levels attributable solely to their generation by mutation, as documented with independent FMDV genetic markers, and with HIV-1 in vivo.[67][68][70][71] Thus, memory is a history-dependent, collective property of the quasispecies that confers a selective advantage to respond to environmental changes previously experienced by the same evolutionary lineage. It can be manifested only if the mutant spectrum maintains its completeness, since memory is lost when the population undergoes a bottleneck event that excludes minorities. A relevant example of the consequences of memory occurs in antiviral pharmacology with the administration for a second time of the same or a related antiviral agent (capable of evoking shared resistance mutations) used in a previous treatment. The second intervention may face inhibitor-resistant memory genomes from the earlier treatment, thus contributing to virus escape.[67] This is an aspect that has not received adequate attention in the planning of antiviral interventions for patients who fail a first treatment and have to be subjected to a second treatment.
Intra-mutant spectrum interactions for interference, complementation or cooperation
Individual genomes surrounded by a cloud of related mutants can be either suppressed to be kept at low frequency, or helped to be maintained in the population. The two alternative fates are dependent on several factors, one being the surrounding mutant spectrum in those steps of the infectious cycle in which an effective competition among variants is established, for example within replication complexes. This important concept was first derived theoretically,[10][72] and then approached experimentally with several viruses. In an early study, Juan Carlos de la Torre and John Holland described suppression of high fitness VSV by mutant spectra of inferior fitness.[10] Suppressive effects have since been documented with standard and mutagenized viral populations. Some examples are:
- Suppression of high fitness antigenic variants of FMDV by low fitness antibody-escape mutants.[73]
- Suppression of virulent poliovirus (PV) by attenuated virus in poliovirus vaccines.[63]
- Suppression of pathogenic lymphocytic choriomengitis virus (LCMV) (that cause growth hormone deficiency in mice) by non-pathogenic LCMV variants.[74]
- Suppression of FMDV by a mutagenized FMDV population.[75]
- Suppression of FMDV by capsid and polymerase FMDV mutants.[76]
- Suppression of drug-resistant viral mutants during antiviral therapy.[77][78]
Opposite to suppression is maintenance of a mutant either by a favorable position in a fitness landscape or by interactions of complementation or cooperation with members of the mutant spectrum. The position in a fitness landscape influences vulnerability to mutations, as popularized with the terms “advantage of the flattest” or “survival of the flattest”, indicating that a variant located at the top of a sharp fitness peak has higher probability to decrease fitness as a result of new mutations than the same variant located at a fitness plateau.[2][79][80] Survival of the flattest has been also proposed as an ingredient in some models of the error threshold.[81]
Collective behavior of viruses was documented with mutant RNA viruses resistant to nucleotide analogues. The study of this class of mutants has been instrumental for the understanding of the molecular basis of template copying fidelity, and the consequences of fidelity alterations in the adaptive capacity and pathogenic potential of RNA viruses.[82][83][84] In the first mutant studied, amino acid substitution G46S in the PV polymerase resulted in about four-fold increase in template-copying fidelity. This modification reduced PV adaptability and infective potential in vivo.[82][83] The mutant in isolation did not replicate efficiently in the brain of susceptible mice, but it did when its mutant spectrum was broadened by 5-fluorouracil mutagenesis or when it was co-inoculated with wild type PV.[83]
Complementation (often occurring when a functional protein encoded by a set of genomes is used by another set of genomes whose encoded protein is not functional) may underlie some collective responses of quasispecies such as fitness of individuals isolated from a population being inferior to fitness of the population.[13][28] Complementation was described between two truncated FMDV genomic forms.[85] The genomes with internal deletions became detectable upon high multiplicity passage of a clonal population of standard FMDV, a virus with a monopartite single stranded RNA genome. Infectivity was generated by complementation of the two truncated forms, in absence of standard, full length FMDV genomes. For complementation to be effective, prior exploration of sequence space through point mutations was a requirement.[86] The system underwent a remarkable evolutionary transition akin to genome segmentation. Drastic genetic lesions in viral genomes are difficult to observe unless a mechanism such as complementation comes into the rescue of the deviant genomes. Additional examples of complementation among RNA viruses have been reported.[87][88][89][40][42] Complementation is a means to maintain defective genomes at detectable frequencies in viral populations.
A distinction has been made between complementation and cooperation, in which two different genomes give rise to a new phenotype through the interaction between two variant proteins.[90] An example of cooperation was characterized during studies with measles virus on membrane fusion which is essential for virus entry into cells. For this virus fusion is mediated by two proteins termed H and F. A truncated H was deficient in cell fusion but the activity was regained when the truncated H was accompanied by two forms of F but not one of the forms individually.[90]
Therefore, complementation, cooperation, interference and suppression can emerge from interactions among components of mutant spectra that have their origin in random mutations. Selection acts on whatever sets of mutants can provide a useful trait, to turn random occurrences into biological meaning.
Bottlenecks
A means to interrupt the participation of individual genomes in interactions with their mutant spectrum is for the quasispecies swarm to undergo drastic reductions in population size that isolate one or few individual genomes from their surroundings. Such reductions are termed bottlenecks, and they have an important participation in shaping evolutionary lineages for all kinds of organisms, and also for viruses. They occur frequently not only upon host-to host transmission but also inside infected hosts,[91][92][93] and they can perturb positive and negative selection events in processes that are difficult to identify and characterize.
Drastic bottleneck events have been reproduced with laboratory populations of viruses in the form of plaque-to-plaque transfers.[94][95] This design served to verify experimentally the operation of Müller’s ratchet, or fitness decrease by the irreversible incorporation of mutations in asexual organisms in absence of compensatory mechanisms.[96] The serial bottleneck transfers unveiled the presence rare mutations, not seen in standard laboratory or natural viral populations. In absence of forced bottleneck events, such rare mutations would be lost by negative selection because of their fitness cost.[97] The investigation of how FMDV clones debilitated by Müller’s ratchet regained replicative fitness revealed several alternative molecular pathways for fitness recovery.[98] The implications of this observation went largely unnoticed until recent results with hepatitis C virus (HCV) have also suggested the accessibility of multiple pathways for fitness gain.[58][60] Also, extensive passage of a biological clone of FMDV in BHK-21 cells conferred the capacity to infect several human cell lines in addition to the expected fitness increase for multiplication in BHK-21 cells.[99] Thus, several lines of evidence suggest that fitness gain in a specific environment may paradoxically broaden the phenotypic potential of a virus. It will be interesting to investigate whether focused adaptation of other viruses to a specific environment may also entail a broadening of diversity, with many phenotypic variants attaining similar fitness levels. If generalized, this broadening of phenotypic space would provide a new interpretation of the molecular basis of adaptation, and explain why adaptation to alternative environments may not lead to attenuation.
Deprivation of an individual virus from possible suppression, complementation or cooperation, may represent a liberation to initiate a new evolutionary process, or a condemnation to extinction. If liberated from suppression, the isolated genome must replicate and be able to reconstruct a mutant cloud to regain adaptive capability. This has led to the suggestion that high mutation rates evolved to allow such mutant spectrum recovery following bottlenecks. Other models attribute high mutation rates to adaptive optimization independent of bottlenecks, or to a mechanistic consequence of rapid replication.[56] Whatever their ultimate origins, high mutation rates serve the purpose of adaptation in multiple circumstances, not only following bottlenecks. A founder virus can introduce a different phenotype for the ensuing evolution. Evolution of viruses in nature and as disease agents can be viewed as succession of mutant spectrum alterations, subjected to expansions and reductions of population size in a continuous interplay of positive and negative selection and random drift. While short-term (for example, intra-host) evolution is observable and measurable, viruses may appear to be relatively static in the long term for decades (as seen with antigenic variants of FMDV [100]) or longer. Intra-host evolution is generally more rapid than inter-host evolution, as documented with viruses[8] and other biological systems.[101] Apparent invariance may be the result of selection for long-term survival of populations that have previously frenziedly tested evolutionary outcomes in short-term processes.[56]
Viral disease
Soon after quasispecies was evidenced for viruses, some medical implications were made explicit.[18][102] Several specific and general points below.[8][41][64][26]
- High mutation rates and population heterogeneity endow viruses with the potential to escape immune pressures (including those due to vaccination) and antiviral inhibitors used in therapy. It is an open question if vaccination can promote long-term evolution of antigenic determinants.
- Attenuated RNA virus vaccines can revert to virulent forms. RNA viruses released in nature for pest control purposes can mutate to new phenotypes.
- Virus attenuation and virulence is dependent on viral genetic traits. Variant forms of a given virus may display increased virulence or atypical disease.
- Components of a mutant spectrum can exhibit a different cell tropism or host range than most genomes in the same population, with implications for the emergence and re-emergence of viral disease.
- Viral pathogenesis is influenced by microevolutionary processes in which some viral subpopulations are replaced by others to persist or to invade new cell types, tissues or organs.
- The larger the actively replicating (effective) population size and the replication rate, the most effective is exploration of sequence space for phenotypic expansions that favor survival and persistence.
- There is a connection between four parameters that characterize viruses during infection processes: replication rate (the rate at which viral RNA or DNA is synthesized intracellularly for viral progeny production), viral load (the total amount of virus quantified in an infected host or host compartment), genetic heterogeneity, and replicative fitness (the yield of infectious particles that can contribute to the next generation). They can influence disease progression, and any of them can be targeted for disease control.
In all interactions conductive to disease, the host cells individually and as groups in tissues and organs play decisive roles. The consequences of a viral infection are always host-dependent. However, the virus itself poses a major challenge that a deeper understanding of quasispecies dynamics is helping to confront.[26]
Antiviral strategies
There is an increasing perception that Darwinian principles should assist in the planning of antiviral designs.[103] The aim of vaccination is to evoke a protective response that either prevents virus replication or disease. The aim of an antiviral pharmacological intervention is to inhibit virus replication to provide the immune system with an opportunity to clear the virus. Expressed simply, the direct danger for vaccination and treatment is that the virus can escape through selection of mutants resistant to vaccine-triggered defense components or to the externally administered inhibitors. This has led to several proposals to confront viral disease, that can be summarized below.[56]
Vaccine exposure of multiple B cell and T cell epitopes
Vaccines should include repertoires of B cell and T cell epitopes to evoke an ample immune response. The broad response should minimize selection of escape mutants that may be present as minority components in mutant spectra, as repeatedly documented experimentally.[8][20][42][67] With the current types of available vaccines, those that best comply with the multiple epitope requirement are, in the order of expected efficacy to confer protection against highly variable viruses: attenuated > inactivated whole virus > several expressed proteins > one expressed protein > multiple synthetic peptide antigens > single peptide antigen. The scarcity of effective synthetic vaccines for RNA viral pathogens despite huge scientific and economic efforts is a reflection of the underlying problems.
Antiviral agents used in combination
Antiviral monotherapy (use of a single antiviral agent) is to be avoided. The following recommendations have been made and in some cases successfully implemented:
- Inhibitors used in combination should target different viral gene products.
- Splitting a treatment into two steps: first an induction regimen, and a second maintenance regimen. Drugs administered in the two steps should be different.
- Targeting of cellular functions needed for the virus life cycle.
- Use of innate immune response-stimulating drugs (for example, inhibitors of enzymes involved in pyrimidine biosynthesis).
- Combined use of immunotherapy and chemotherapy.
- Lethal mutagenesis or virus extinction by excess of mutations introduced during viral replication.
These strategies have as their main objective to avoid selection of treatment-escape mutants by multiple selective constraints that cannot be surmounted by the virus.[56][104] Control is effective either because exploration of sequence space cannot reach the required multiple mutations (even when recombination is available) or because the multiple mutations inflict a severe fitness cost.[104] Vaccines exposing multiple epitopes and combination therapies follow the same strategy whose aim is to limit possible escape routes to viral quasispecies in the face of the suppressive constraint.
Lethal mutagenesis
Lethal mutagenesis is the process of virus extinction at the error rate at which a virus can no longer maintain its genetic information.[8][26][42][56][60][81][105][106] Application of lethal mutagenesis as an antiviral strategy deserves attention in the context of the present article because its origins lie in quasispecies theory, in the form of the error threshold relationship. Both the error threshold and lethal mutagenesis are highly fitness landscape-dependent, but both can occur in complex fitness landscapes as those pertinent to viral populations.[81] The term lethal mutagenesis was coined by Lawerence Loeb and colleagues,[105] and it is now widely used to describe the antiviral activity of base and nucleoside analogues that increase the viral mutation rate. Although several models have been proposed to account for virus extinction by excess mutations,[81] an extension of the violation of the error threshold stands as a likely mechanism.[107][106] Interestingly, some antiviral agents licensed for human use, initially thought to act only as inhibitors of viral replication, may actually exert their antviral activity against some RNA viruses at least partially by lethal mutagenesis. This is the case of favipiravir (T-705; 6-fluoro-3-hydroxy-2-pirazinecarboxamide) and ribavirin (1-β-D-ribofuranosyl-1-H-1,2,4-triazole-3-carboxamide) that are currently being intensively investigated as lethal mutagens.[106]
Defense mechanisms based on genome modification of invading genetic parasites such as editing cellular activities that are recruited as part of the innate immune response (ADAR, APOBEC, RIP, etc.)[108] represent a natural counterpart of the principle utilized by lethal mutagenesis. Applicability to pathogenic cellular elements is a real possibility, and lethal mutagenesis to control tumor cells is an active field of investigation.[109][110] Thus, the recognition of quasispecies dynamics has suggested some fundamental guidelines for disease prevention and control that are gradually permeating clinical practice. This is in line with the recognized need to apply Darwinian principles to the control of infectious disease.
Error threshold
This may be defined as “The inability of a genetic element to be maintained in a population as the fidelity of its replication machinery decreases beyond a certain threshold value”.[111]
In theory, if the mutation rate was sufficiently high, the viral population would not be able to maintain the genotype with the highest fitness, and therefore the ability of the population to adapt to its environment would be compromised. A practical application of this dynamic is in antiviral drugs employing lethal mutagenesis. For example, increased doses of the mutagen Ribavirin reduces the infectivity of Poliovirus.[112]
However, these models assume that only the mutations that occur in the fittest sequence are deleterious, and furthermore that they are non-lethal. It has been argued that, if we take into account the deleterious effect of mutations on the population of variants and the fact that many mutations are lethal, then the Error Threshold disappears, i.e. the fittest sequence always maintains itself.[113][111][114] Empirical data on the effect of mutations in viruses is rare, but appears to correspond with this scenario.[115]
Possible evolutionary consequences
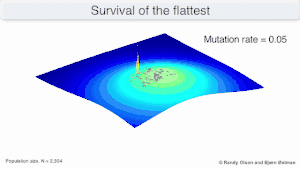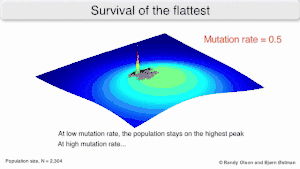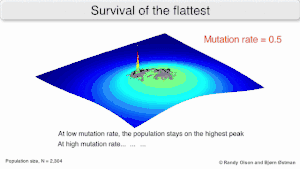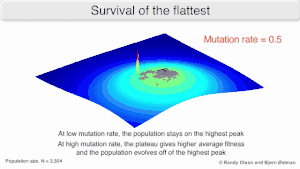
Mutational robustness
The long-term evolution of the virus may be influenced in that it may be a better evolutionarily stable strategy to generate a broad quasispecies with members of approximately equal fitness than to have a sharply defined 'most fit' single genotype (with mutational neighbours substantially less fit). This has been called 'survival of the flattest' - referring to the fitness profiles of the two strategies respectively.[2]
Over the long-term, a flatter fitness profile might better allow a quasispecies to exploit changes in selection pressure, analogous to the way sexual organisms use recombination to preserve diversity in a population. At least in simulations, a slower replicator can be shown to be able to outcompete a faster one in cases where it is more robust and the mutation rate is high.[1]
However, whether mutational robustness evolved or is intrinsic to genetic systems is unconfirmed, because the basic mechanism behind robustness would depend upon the peculiarities of each system.[3]
Cooperation
Experimental manipulation of poliovirus to give them a higher-fidelity polymerase – and hence reduce their mutation rate – showed these variants to have lower pathogenicity than wild-type sequences.[83] Pathogenicity could then be restored by mutagen application. This was interpreted to mean lower mutation rates had reduced the adaptability (or breadth) of the quasispecies. The mutant viruses extracted from brain tissue were not themselves pathogenic, and the authors speculate that there may be complementation between variant members of the quasispecies that could enable viruses to colonize different host tissues and systems.
References
![]() This article was adapted from the following source under a CC BY 4.0 license (0200) (reviewer reports): "Viral quasispecies". PLOS Genetics. 15 (10): e1008271. 17 October 2019. doi:10.1371/JOURNAL.PGEN.1008271. ISSN 1553-7390. PMC 6797082. PMID 31622336. Wikidata Q86320171.
This article was adapted from the following source under a CC BY 4.0 license (0200) (reviewer reports): "Viral quasispecies". PLOS Genetics. 15 (10): e1008271. 17 October 2019. doi:10.1371/JOURNAL.PGEN.1008271. ISSN 1553-7390. PMC 6797082. PMID 31622336. Wikidata Q86320171.
- van Nimwegen E, Crutchfield JP, Huynen M (August 1999). "Neutral evolution of mutational robustness". Proceedings of the National Academy of Sciences of the United States of America. 96 (17): 9716–20. arXiv:adap-org/9903006. Bibcode:1999PNAS...96.9716V. doi:10.1073/pnas.96.17.9716. PMC 22276. PMID 10449760.
- Wilke CO, Wang JL, Ofria C, Lenski RE, Adami C (July 2001). "Evolution of digital organisms at high mutation rates leads to survival of the flattest". Nature. 412 (6844): 331–3. Bibcode:2001Natur.412..331W. doi:10.1038/35085569. PMID 11460163.
- Elena SF, Agudelo-Romero P, Carrasco P, Codoñer FM, Martín S, Torres-Barceló C, Sanjuán R (May 2008). "Experimental evolution of plant RNA viruses". Heredity. 100 (5): 478–83. doi:10.1038/sj.hdy.6801088. PMC 7094686. PMID 18253158.
- Eigen M, McCaskill J, Schuster P (1988). "Molecular Quasi-Species". Journal of Physical Chemistry. 92 (24): 6881–6891. doi:10.1021/j100335a010. hdl:11858/00-001M-0000-002C-84A7-C. S2CID 96727272.
- Nowak MA (April 1992). "What is a quasispecies?". Trends in Ecology & Evolution. 7 (4): 118–21. doi:10.1016/0169-5347(92)90145-2. PMID 21235976.
- Drake JW, Holland JJ (November 1999). "Mutation rates among RNA viruses". Proceedings of the National Academy of Sciences of the United States of America. 96 (24): 13910–3. Bibcode:1999PNAS...9613910D. doi:10.1073/pnas.96.24.13910. PMC 24164. PMID 10570172.
- Eigen, Manfred; Schuster, Peter (1979). "The Hypercycle". Naturwissenschaften. 65 (1): 7–41. doi:10.1007/bf00420631. ISSN 0028-1042.
- Domingo E, Sheldon J, Perales C (June 2012). "Viral quasispecies evolution". Microbiology and Molecular Biology Reviews. 76 (2): 159–216. doi:10.1128/MMBR.05023-11. PMC 3372249. PMID 22688811.
- Eigen M (October 1971). "Selforganization of matter and the evolution of biological macromolecules". Die Naturwissenschaften. 58 (10): 465–523. Bibcode:1971NW.....58..465E. doi:10.1007/bf00623322. PMID 4942363.
- Swetina J, Schuster P (December 1982). "Self-replication with errors. A model for polynucleotide replication". Biophysical Chemistry. 16 (4): 329–45. doi:10.1016/0301-4622(82)87037-3. PMID 7159681.
- Fornés J, Tomás Lázaro J, Alarcón T, Elena SF, Sardanyés J (January 2019). "Viral replication modes in single-peak fitness landscapes: A dynamical systems analysis". Journal of Theoretical Biology. 460: 170–183. doi:10.1016/j.jtbi.2018.10.007. hdl:2072/378023. PMID 30300648.
- Schuster P (2016). Quasispecies on Fitness Landscapes. Current Topics in Microbiology and Immunology. 392. Springer International Publishing. pp. 61–120. doi:10.1007/82_2015_469. ISBN 9783319238975. PMID 26597856.
- Domingo E, Sabo D, Taniguchi T, Weissmann C (April 1978). "Nucleotide sequence heterogeneity of an RNA phage population". Cell. 13 (4): 735–44. doi:10.1016/0092-8674(78)90223-4. PMID 657273.
- Duarte EA, Novella IS, Ledesma S, Clarke DK, Moya A, Elena SF, et al. (July 1994). "Subclonal components of consensus fitness in an RNA virus clone". Journal of Virology. 68 (7): 4295–301. doi:10.1128/JVI.68.7.4295-4301.1994. PMC 236352. PMID 8207804.
- Domingo E, Brun A, Nuñez JI, Cristina J, Briones C, Escarmís C (2006-05-10). "Genomics of Viruses". Pathogenomics. Wiley-VCH Verlag GmbH & Co. KGaA: 367–388. doi:10.1002/352760801x.ch17. ISBN 9783527608010.
- Batschelet E, Domingo E, Weissmann C (January 1976). "The proportion of revertant and mutant phage in a growing population, as a function of mutation and growth rate". Gene. 1 (1): 27–32. doi:10.1016/0378-1119(76)90004-4. PMID 1052321.
- Bradwell K, Combe M, Domingo-Calap P, Sanjuán R (September 2013). "Correlation between mutation rate and genome size in riboviruses: mutation rate of bacteriophage Qβ". Genetics. 195 (1): 243–51. doi:10.1534/genetics.113.154963. PMC 3761305. PMID 23852383.
- Holland J, Spindler K, Horodyski F, Grabau E, Nichol S, VandePol S (March 1982). "Rapid evolution of RNA genomes". Science. 215 (4540): 1577–85. Bibcode:1982Sci...215.1577H. doi:10.1126/science.7041255. PMID 7041255.
- Domingo E, Martínez-Salas E, Sobrino F, de la Torre JC, Portela A, Ortín J, et al. (January 1985). "The quasispecies (extremely heterogeneous) nature of viral RNA genome populations: biological relevance--a review". Gene. 40 (1): 1–8. doi:10.1016/0378-1119(85)90017-4. PMID 3912262.
- Domingo E, Holland JJ, Ahlquist P (1988). Domingo E, Holland JJ, Ahlquist P (eds.). RNA Genetics. doi:10.1201/9781351076432. ISBN 9781351076432.
- Holland JJ (2006). "Transitions in understanding of RNA viruses: a historical perspective". Quasispecies: Concept and Implications for Virology. Current Topics in Microbiology and Immunology. 299. Springer-Verlag. pp. 371–401. doi:10.1007/3-540-26397-7_14. ISBN 3540263950. PMID 16568907.
- Meyerhans A, Cheynier R, Albert J, Seth M, Kwok S, Sninsky J, et al. (September 1989). "Temporal fluctuations in HIV quasispecies in vivo are not reflected by sequential HIV isolations". Cell. 58 (5): 901–10. doi:10.1016/0092-8674(89)90942-2. PMID 2550139.
- Farci P (November 2011). "New insights into the HCV quasispecies and compartmentalization". Seminars in Liver Disease. 31 (4): 356–74. doi:10.1055/s-0031-1297925. PMID 22189976.
- Eigen M, Schuster P (November 1977). "The hypercycle. A principle of natural self-organization. Part A: Emergence of the hypercycle". Die Naturwissenschaften. 64 (11): 541–65. doi:10.1007/bf00450633. PMID 593400.
- Saakian DB, Hu CK (2016). "Mathematical Models of Quasi-Species Theory and Exact Results for the Dynamics". Current Topics in Microbiology and Immunology. Springer International Publishing. 392: 121–39. doi:10.1007/82_2015_471. ISBN 9783319238975. PMID 26342705.
- Domingo E, Schuster P, eds. (2016). Quasispecies: From Theory to Experimental Systems. Current Topics in Microbiology and Immunology. 392. doi:10.1007/978-3-319-23898-2. ISBN 978-3-319-23897-5.
- Steinhauer DA, Domingo E, Holland JJ (December 1992). "Lack of evidence for proofreading mechanisms associated with an RNA virus polymerase". Gene. 122 (2): 281–8. doi:10.1016/0378-1119(92)90216-c. PMID 1336756.
- Bernad A, Blanco L, Lázaro JM, Martín G, Salas M (October 1989). "A conserved 3'----5' exonuclease active site in prokaryotic and eukaryotic DNA polymerases". Cell. 59 (1): 219–28. doi:10.1016/0092-8674(89)90883-0. PMID 2790959.
- Eckerle LD, Lu X, Sperry SM, Choi L, Denison MR (November 2007). "High fidelity of murine hepatitis virus replication is decreased in nsp14 exoribonuclease mutants". Journal of Virology. 81 (22): 12135–44. doi:10.1128/jvi.01296-07. PMC 2169014. PMID 17804504.
- Nagy PD, Carpenter CD, Simon AE (February 1997). "A novel 3'-end repair mechanism in an RNA virus". Proceedings of the National Academy of Sciences of the United States of America. 94 (4): 1113–8. Bibcode:1997PNAS...94.1113N. doi:10.1073/pnas.94.4.1113. PMC 19753. PMID 9037015.
- Bakhanashvili M (April 2001). "Exonucleolytic proofreading by p53 protein". European Journal of Biochemistry. 268 (7): 2047–54. doi:10.1046/j.1432-1327.2001.02075.x. PMID 11277927.
- Smith EC, Denison MR (2013-12-05). "Coronaviruses as DNA wannabes: a new model for the regulation of RNA virus replication fidelity". PLOS Pathogens. 9 (12): e1003760. doi:10.1371/journal.ppat.1003760. PMC 3857799. PMID 24348241.
- Smith EC, Sexton NR, Denison MR (November 2014). "Thinking Outside the Triangle: Replication Fidelity of the Largest RNA Viruses". Annual Review of Virology. 1 (1): 111–32. doi:10.1146/annurev-virology-031413-085507. PMID 26958717.
- Wagner N, Atsmon-Raz Y, Ashkenasy G (2016). "Theoretical Models of Generalized Quasispecies". Current Topics in Microbiology and Immunology. Springer International Publishing. 392: 141–59. doi:10.1007/82_2015_456. ISBN 9783319238975. PMID 26373410.
- Schmidt TT, Reyes G, Gries K, Ceylan CÜ, Sharma S, Meurer M, et al. (May 2017). "GLN3 inactivation cause imbalanced dNTP pools and increased mutagenesis". Proceedings of the National Academy of Sciences of the United States of America. 114 (22): E4442–E4451. doi:10.1073/pnas.1618714114. PMC 5465912. PMID 28416670.
- Takahashi K, Sekizuka T, Fukumoto H, Nakamichi K, Suzuki T, Sato Y, et al. (January 2017). "Deep-Sequence Identification and Role in Virus Replication of a JC Virus Quasispecies in Patients with Progressive Multifocal Leukoencephalopathy". Journal of Virology. 91 (1). doi:10.1128/jvi.01335-16. PMC 5165223. PMID 27795410.
- Domingo-Calap P, Schubert B, Joly M, Solis M, Untrau M, Carapito R, et al. (October 2018). "An unusually high substitution rate in transplant-associated BK polyomavirus in vivo is further concentrated in HLA-C-bound viral peptides". PLOS Pathogens. 14 (10): e1007368. doi:10.1371/journal.ppat.1007368. PMC 6207329. PMID 30335851.
- Sánchez-Campos S, Domínguez-Huerta G, Díaz-Martínez L, Tomás DM, Navas-Castillo J, Moriones E, Grande-Pérez A (2018-07-02). "Differential Shape of Geminivirus Mutant Spectra Across Cultivated and Wild Hosts With Invariant Viral Consensus Sequences". Frontiers in Plant Science. 9: 932. doi:10.3389/fpls.2018.00932. PMC 6036239. PMID 30013589.
- Donohue RC, Pfaller CK, Cattaneo R (February 2019). "Cyclical adaptation of measles virus quasispecies to epithelial and lymphocytic cells: To V, or not to V". PLOS Pathogens. 15 (2): e1007605. doi:10.1371/journal.ppat.1007605. PMC 6395005. PMID 30768648.
- Agol VI, Gmyl AP (June 2018). "Emergency Services of Viral RNAs: Repair and Remodeling". Microbiology and Molecular Biology Reviews. 82 (2): e00067-1. doi:10.1128/mmbr.00067-17. PMC 5968460. PMID 29540453.
- Figlerowicz, Magdalena; Alejska, Magdalena; Kurzynska-Kokorniak, Anna; Figlerowicz, Marek (2003-09-23). "Genetic Variability: The Key Problem in the Prevention and Therapy of RNA-Based Virus Infections". ChemInform. 34 (38). doi:10.1002/chin.200338243. ISSN 0931-7597.
- Domingo E, Perales C (May 2018). "Quasispecies and virus". European Biophysics Journal. 47 (4): 443–457. doi:10.1007/s00249-018-1282-6. PMID 29397419.
- Sanjuán R, Domingo-Calap P (December 2016). "Mechanisms of viral mutation". Cellular and Molecular Life Sciences. 73 (23): 4433–4448. doi:10.1007/s00018-016-2299-6. PMC 5075021. PMID 27392606.
- Lauring AS, Andino R (July 2010). "Quasispecies theory and the behavior of RNA viruses". PLOS Pathogens. 6 (7): e1001005. doi:10.1371/journal.ppat.1001005. PMC 2908548. PMID 20661479.
- van Boheemen S, Tas A, Anvar SY, van Grootveld R, Albulescu IC, Bauer MP, et al. (May 2017). "Quasispecies composition and evolution of a typical Zika virus clinical isolate from Suriname". Scientific Reports. 7 (1): 2368. Bibcode:2017NatSR...7.2368V. doi:10.1038/s41598-017-02652-w. PMC 5443807. PMID 28539654.
- Vlok M, Lang AS, Suttle CA (April 2019). "Marine RNA Virus Quasispecies Are Distributed throughout the Oceans". mSphere. 4 (2): e00157-19. doi:10.1128/mspheredirect.00157-19. PMC 6449609. PMID 30944212.
- Hirose Y, Onuki M, Tenjimbayashi Y, Mori S, Ishii Y, Takeuchi T, et al. (June 2018). "Within-Host Variations of Human Papillomavirus Reveal APOBEC Signature Mutagenesis in the Viral Genome". Journal of Virology. 92 (12): e00017-18. doi:10.1128/jvi.00017-18. PMC 5974501. PMID 29593040.
- Gisder S, Möckel N, Eisenhardt D, Genersch E (December 2018). "In vivo evolution of viral virulence: switching of deformed wing virus between hosts results in virulence changes and sequence shifts". Environmental Microbiology. 20 (12): 4612–4628. doi:10.1111/1462-2920.14481. PMID 30452113.
- Baccam P, Thompson RJ, Fedrigo O, Carpenter S, Cornette JL (January 2001). "PAQ: Partition Analysis of Quasispecies". Bioinformatics. 17 (1): 16–22. doi:10.1093/bioinformatics/17.1.16. PMID 11222259.
- Skums P, Zelikovsky A, Singh R, Gussler W, Dimitrova Z, Knyazev S, et al. (January 2018). "QUENTIN: reconstruction of disease transmissions from viral quasispecies genomic data". Bioinformatics. 34 (1): 163–170. doi:10.1093/bioinformatics/btx402. PMC 6355096. PMID 29304222.
- Lowry K, Woodman A, Cook J, Evans DJ (June 2014). "Recombination in enteroviruses is a biphasic replicative process involving the generation of greater-than genome length 'imprecise' intermediates". PLOS Pathogens. 10 (6): e1004191. doi:10.1371/journal.ppat.1004191. PMC 4055744. PMID 24945141.
- Xiao Y, Rouzine IM, Bianco S, Acevedo A, Goldstein EF, Farkov M, et al. (September 2017). "RNA Recombination Enhances Adaptability and Is Required for Virus Spread and Virulence". Cell Host & Microbe. 22 (3): 420. doi:10.1016/j.chom.2017.08.006. PMC 5807061. PMID 28910639.
- Tibayrenc M, Ayala FJ (November 2012). "Reproductive clonality of pathogens: a perspective on pathogenic viruses, bacteria, fungi, and parasitic protozoa". Proceedings of the National Academy of Sciences of the United States of America. 109 (48): E3305-13. doi:10.1073/pnas.1212452109. PMC 3511763. PMID 22949662.
- Perales C, Moreno E, Domingo E (July 2015). "Clonality and intracellular polyploidy in virus evolution and pathogenesis". Proceedings of the National Academy of Sciences of the United States of America. 112 (29): 8887–92. Bibcode:2015PNAS..112.8887P. doi:10.1073/pnas.1501715112. PMC 4517279. PMID 26195777.
- Villarreal LP, Witzany G (November 2013). "Rethinking quasispecies theory: From fittest type to cooperative consortia". World Journal of Biological Chemistry. 4 (4): 79–90. doi:10.4331/wjbc.v4.i4.79. PMC 3856310. PMID 24340131.
- Domingo E (September 2015). Virus as populations: composition, complexity, dynamics, and biological implications. Academic Press. ISBN 978-0-12-800837-9.
- Acevedo A, Brodsky L, Andino R (January 2014). "Mutational and fitness landscapes of an RNA virus revealed through population sequencing". Nature. 505 (7485): 686–90. Bibcode:2014Natur.505..686A. doi:10.1038/nature12861. PMC 4111796. PMID 24284629.
- Moreno E, Gallego I, Gregori J, Lucía-Sanz A, Soria ME, Castro V, et al. (May 2017). "Internal Disequilibria and Phenotypic Diversification during Replication of Hepatitis C Virus in a Noncoevolving Cellular Environment". Journal of Virology. 91 (10). doi:10.1128/jvi.02505-16. PMC 5411618. PMID 28275194.
- Ojosnegros S, Beerenwinkel N, Antal T, Nowak MA, Escarmís C, Domingo E (February 2010). "Competition-colonization dynamics in an RNA virus". Proceedings of the National Academy of Sciences of the United States of America. 107 (5): 2108–12. Bibcode:2010PNAS..107.2108O. doi:10.1073/pnas.0909787107. PMC 2836666. PMID 20080701.
- Gallego I, Gregori J, Soria ME, García-Crespo C, García-Álvarez M, Gómez-González A, et al. (October 2018). "Resistance of high fitness hepatitis C virus to lethal mutagenesis". Virology. 523: 100–109. doi:10.1016/j.virol.2018.07.030. PMID 30107298.
- Holland JJ, De La Torre JC, Steinhauer DA (1992). "RNA virus populations as quasispecies". Current Topics in Microbiology and Immunology. 176: 1–20. doi:10.1007/978-3-642-77011-1_1. ISBN 9783642770111. OCLC 851813241. PMID 1600747.
- García-Arriaza J, Ojosnegros S, Dávila M, Domingo E, Escarmís C (July 2006). "Dynamics of mutation and recombination in a replicating population of complementing, defective viral genomes". Journal of Molecular Biology. 360 (3): 558–72. doi:10.1016/j.jmb.2006.05.027. PMID 16797586.
- Chumakov KM, Powers LB, Noonan KE, Roninson IB, Levenbook IS (January 1991). "Correlation between amount of virus with altered nucleotide sequence and the monkey test for acceptability of oral poliovirus vaccine". Proceedings of the National Academy of Sciences of the United States of America. 88 (1): 199–203. Bibcode:1991PNAS...88..199C. doi:10.1073/pnas.88.1.199. PMC 50777. PMID 1846038.
- Holland JJ (1992). Genetic Diversity of RNA Viruses. Berlin, Heidelberg: Springer Berlin Heidelberg. ISBN 9783642770111. OCLC 851813241.
- Perales C (October 2018). "Quasispecies dynamics and clinical significance of hepatitis C virus (HCV) antiviral resistance". International Journal of Antimicrobial Agents: 105562. doi:10.1016/j.ijantimicag.2018.10.005. PMID 30315919.
- Martín V, Domingo E (August 2008). "Influence of the mutant spectrum in viral evolution: focused selection of antigenic variants in a reconstructed viral quasispecies". Molecular Biology and Evolution. 25 (8): 1544–54. doi:10.1093/molbev/msn099. PMID 18436553.
- Briones C, Domingo E (2008). "Minority report: hidden memory genomes in HIV-1 quasispecies and possible clinical implications". AIDS Reviews. 10 (2): 93–109. PMID 18615120.
- Ruiz-Jarabo CM, Arias A, Baranowski E, Escarmís C, Domingo E (April 2000). "Memory in viral quasispecies". Journal of Virology. 74 (8): 3543–7. doi:10.1128/jvi.74.8.3543-3547.2000. PMC 111862. PMID 10729128.
- Farber DL, Netea MG, Radbruch A, Rajewsky K, Zinkernagel RM (February 2016). "Immunological memory: lessons from the past and a look to the future". Nature Reviews. Immunology. 16 (2): 124–8. doi:10.1038/nri.2016.13. PMID 26831526.
- Briones C, Domingo E, Molina-París C (August 2003). "Memory in retroviral quasispecies: experimental evidence and theoretical model for human immunodeficiency virus". Journal of Molecular Biology. 331 (1): 213–29. doi:10.1016/s0022-2836(03)00661-2. PMC 7173031. PMID 12875847.
- Arias A, Ruiz-Jarabo CM, Escarmís C, Domingo E (May 2004). "Fitness increase of memory genomes in a viral quasispecies". Journal of Molecular Biology. 339 (2): 405–12. doi:10.1016/j.jmb.2004.03.061. PMID 15136042.
- Eigen M, Biebricher CK (1988). Sequence Space and Quasispecies Distribution. RNA Genetics. CRC Press. pp. 211–245. doi:10.1201/9781351076449-12. ISBN 9781351076449.
- Borrego B, Novella IS, Giralt E, Andreu D, Domingo E (October 1993). "Distinct repertoire of antigenic variants of foot-and-mouth disease virus in the presence or absence of immune selection". Journal of Virology. 67 (10): 6071–9. doi:10.1128/JVI.67.10.6071-6079.1993. PMC 238028. PMID 7690417.
- Teng MN, Oldstone MB, de la Torre JC (September 1996). "Suppression of lymphocytic choriomeningitis virus--induced growth hormone deficiency syndrome by disease-negative virus variants". Virology. 223 (1): 113–9. doi:10.1006/viro.1996.0460. PMID 8806545.
- González-López C, Arias A, Pariente N, Gómez-Mariano G, Domingo E (April 2004). "Preextinction viral RNA can interfere with infectivity". Journal of Virology. 78 (7): 3319–24. doi:10.1128/jvi.78.7.3319-3324.2004. PMC 371084. PMID 15016853.
- Perales C, Mateo R, Mateu MG, Domingo E (June 2007). "Insights into RNA virus mutant spectrum and lethal mutagenesis events: replicative interference and complementation by multiple point mutants". Journal of Molecular Biology. 369 (4): 985–1000. doi:10.1016/j.jmb.2007.03.074. PMID 17481660.
- Crowder S, Kirkegaard K (July 2005). "Trans-dominant inhibition of RNA viral replication can slow growth of drug-resistant viruses". Nature Genetics. 37 (7): 701–9. doi:10.1038/ng1583. PMID 15965477.
- Kirkegaard K, van Buuren NJ, Mateo R (October 2016). "My Cousin, My Enemy: quasispecies suppression of drug resistance". Current Opinion in Virology. 20: 106–111. doi:10.1016/j.coviro.2016.09.011. PMC 5298929. PMID 27764731.
- Quer J, Hershey CL, Domingo E, Holland JJ, Novella IS (August 2001). "Contingent neutrality in competing viral populations". Journal of Virology. 75 (16): 7315–20. doi:10.1128/jvi.75.16.7315-7320.2001. PMC 114966. PMID 11462003.
- Codoñer FM, Darós JA, Solé RV, Elena SF (December 2006). "The fittest versus the flattest: experimental confirmation of the quasispecies effect with subviral pathogens". PLOS Pathogens. 2 (12): e136. doi:10.1371/journal.ppat.0020136. PMC 1757203. PMID 17196038.
- Tejero H, Montero F, Nuño JC (2016). "Theories of Lethal Mutagenesis: From Error Catastrophe to Lethal Defection". Current Topics in Microbiology and Immunology. Springer International Publishing. 392: 161–79. doi:10.1007/82_2015_463. ISBN 9783319238975. PMID 26210988.
- Pfeiffer JK, Kirkegaard K (October 2005). "Increased fidelity reduces poliovirus fitness and virulence under selective pressure in mice". PLOS Pathogens. 1 (2): e11. doi:10.1371/journal.ppat.0010011. PMC 1250929. PMID 16220146.
- Vignuzzi M, Stone JK, Arnold JJ, Cameron CE, Andino R (January 2006). "Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population". Nature. 439 (7074): 344–8. Bibcode:2006Natur.439..344V. doi:10.1038/nature04388. PMC 1569948. PMID 16327776.
- Bordería AV, Rozen-Gagnon K, Vignuzzi M (2016). "Fidelity Variants and RNA Quasispecies". Current Topics in Microbiology and Immunology. Springer International Publishing. 392: 303–22. doi:10.1007/82_2015_483. ISBN 9783319238982. PMC 7121553. PMID 26499340.
- García-Arriaza J, Manrubia SC, Toja M, Domingo E, Escarmís C (November 2004). "Evolutionary transition toward defective RNAs that are infectious by complementation". Journal of Virology. 78 (21): 11678–85. doi:10.1128/JVI.78.21.11678-11685.2004. PMC 523252. PMID 15479809.
- Moreno E, Ojosnegros S, García-Arriaza J, Escarmís C, Domingo E, Perales C (May 2014). "Exploration of sequence space as the basis of viral RNA genome segmentation". Proceedings of the National Academy of Sciences of the United States of America. 111 (18): 6678–83. Bibcode:2014PNAS..111.6678M. doi:10.1073/pnas.1323136111. PMC 4020086. PMID 24757055.
- Aaskov J, Buzacott K, Thu HM, Lowry K, Holmes EC (January 2006). "Long-term transmission of defective RNA viruses in humans and Aedes mosquitoes". Science. 311 (5758): 236–8. Bibcode:2006Sci...311..236A. doi:10.1126/science.1115030. PMID 16410525.
- Ciota AT, Ehrbar DJ, Van Slyke GA, Willsey GG, Kramer LD (May 2012). "Cooperative interactions in the West Nile virus mutant swarm". BMC Evolutionary Biology. 12 (1): 58. doi:10.1186/1471-2148-12-58. PMC 3358237. PMID 22541042.
- Xue KS, Hooper KA, Ollodart AR, Dingens AS, Bloom JD (March 2016). "Cooperation between distinct viral variants promotes growth of H3N2 influenza in cell culture". eLife. 5: e13974. doi:10.7554/elife.13974. PMC 4805539. PMID 26978794.
- Shirogane Y, Watanabe S, Yanagi Y (2016). "Cooperative Interaction Within RNA Virus Mutant Spectra". Current Topics in Microbiology and Immunology. Springer International Publishing. 392: 219–29. doi:10.1007/82_2015_461. ISBN 9783319238975. PMID 26162566.
- Pfeiffer JK, Kirkegaard K (April 2006). "Bottleneck-mediated quasispecies restriction during spread of an RNA virus from inoculation site to brain". Proceedings of the National Academy of Sciences of the United States of America. 103 (14): 5520–5. Bibcode:2006PNAS..103.5520P. doi:10.1073/pnas.0600834103. PMC 1414638. PMID 16567621.
- Gutiérrez S, Michalakis Y, Blanc S (October 2012). "Virus population bottlenecks during within-host progression and host-to-host transmission". Current Opinion in Virology. 2 (5): 546–55. doi:10.1016/j.coviro.2012.08.001. PMID 22921636.
- Bull RA, Luciani F, McElroy K, Gaudieri S, Pham ST, Chopra A, et al. (September 2011). "Sequential bottlenecks drive viral evolution in early acute hepatitis C virus infection". PLOS Pathogens. 7 (9): e1002243. doi:10.1371/journal.ppat.1002243. PMC 3164670. PMID 21912520.
- Chao L (November 1990). "Fitness of RNA virus decreased by Muller's ratchet". Nature. 348 (6300): 454–5. Bibcode:1990Natur.348..454C. doi:10.1038/348454a0. PMID 2247152.
- Duarte E, Clarke D, Moya A, Domingo E, Holland J (July 1992). "Rapid fitness losses in mammalian RNA virus clones due to Muller's ratchet". Proceedings of the National Academy of Sciences of the United States of America. 89 (13): 6015–9. Bibcode:1992PNAS...89.6015D. doi:10.1073/pnas.89.13.6015. PMC 402129. PMID 1321432.
- Muller HJ (May 1964). "The relation of recombination to mutational advance". Mutation Research. 106 (1): 2–9. doi:10.1016/0027-5107(64)90047-8. PMID 14195748.
- Escarmís C, Dávila M, Charpentier N, Bracho A, Moya A, Domingo E (November 1996). "Genetic lesions associated with Muller's ratchet in an RNA virus". Journal of Molecular Biology. 264 (2): 255–67. doi:10.1006/jmbi.1996.0639. PMID 8951375.
- Escarmís C, Dávila M, Domingo E (January 1999). "Multiple molecular pathways for fitness recovery of an RNA virus debilitated by operation of Muller's ratchet". Journal of Molecular Biology. 285 (2): 495–505. doi:10.1006/jmbi.1998.2366. PMID 9878424.
- Ruiz-Jarabo CM, Pariente N, Baranowski E, Dávila M, Gómez-Mariano G, Domingo E (August 2004). "Expansion of host-cell tropism of foot-and-mouth disease virus despite replication in a constant environment". The Journal of General Virology. 85 (Pt 8): 2289–97. doi:10.1099/vir.0.80126-0. PMID 15269370.
- Martínez MA, Dopazo J, Hernández J, Mateu MG, Sobrino F, Domingo E, Knowles NJ (June 1992). "Evolution of the capsid protein genes of foot-and-mouth disease virus: antigenic variation without accumulation of amino acid substitutions over six decades". Journal of Virology. 66 (6): 3557–65. doi:10.1128/JVI.66.6.3557-3565.1992. PMC 241137. PMID 1316467.
- Ho SY, Duchêne S, Molak M, Shapiro B (December 2015). "Time-dependent estimates of molecular evolutionary rates: evidence and causes". Molecular Ecology. 24 (24): 6007–12. doi:10.1111/mec.13450. PMID 26769402. S2CID 14433111.
- Domingo E (1989). "RNA virus evolution and the control of viral disease". Progress in Drug Research. Fortschritte der Arzneimittelforschung. Progrès des Recherches Pharmaceutiques. Birkhäuser Basel. 33: 93–133. doi:10.1007/978-3-0348-9146-2_5. ISBN 9783034899253. PMID 2687948.
- Williams PD (February 2010). "Darwinian interventions: taming pathogens through evolutionary ecology". Trends in Parasitology. 26 (2): 83–92. doi:10.1016/j.pt.2009.11.009. PMID 20036799.
- Perales C, Ortega-Prieto AM, Beach NM, Sheldon J, Menéndez-Arias L, Domingo E (2017). "Quasispecies and Drug Resistance". Handbook of Antimicrobial Resistance. Springer New York: 123–147. doi:10.1007/978-1-4939-0694-9_1. ISBN 9781493906932.
- Loeb LA, Essigmann JM, Kazazi F, Zhang J, Rose KD, Mullins JI (February 1999). "Lethal mutagenesis of HIV with mutagenic nucleoside analogs". Proceedings of the National Academy of Sciences of the United States of America. 96 (4): 1492–7. Bibcode:1999PNAS...96.1492L. doi:10.1073/pnas.96.4.1492. PMC 15492. PMID 9990051.
- Perales C, Gallego I, de Ávila AI, Soria ME, Gregori J, Quer J, Domingo E (July 2019). "The increasing impact of lethal mutagenesis of viruses". Future Medicinal Chemistry. 11 (13): 1645–1657. doi:10.4155/fmc-2018-0457. hdl:10261/216260. PMID 31469331.
- Eigen M (October 2002). "Error catastrophe and antiviral strategy". Proceedings of the National Academy of Sciences of the United States of America. 99 (21): 13374–6. Bibcode:2002PNAS...9913374E. doi:10.1073/pnas.212514799. PMC 129678. PMID 12370416.
- Venkatesan S, Rosenthal R, Kanu N, McGranahan N, Bartek J, Quezada SA, et al. (March 2018). "Perspective: APOBEC mutagenesis in drug resistance and immune escape in HIV and cancer evolution". Annals of Oncology. 29 (3): 563–572. doi:10.1093/annonc/mdy003. PMC 5888943. PMID 29324969.
- Fox EJ, Loeb LA (October 2010). "Lethal mutagenesis: targeting the mutator phenotype in cancer". Seminars in Cancer Biology. 20 (5): 353–9. doi:10.1016/j.semcancer.2010.10.005. PMC 3256989. PMID 20934515.
- Loeb LA (June 2011). "Human cancers express mutator phenotypes: origin, consequences and targeting". Nature Reviews. Cancer. 11 (6): 450–7. doi:10.1038/nrc3063. PMC 4007007. PMID 21593786.
- Summers J, Litwin S (January 2006). "Examining the theory of error catastrophe". Journal of Virology. 80 (1): 20–6. doi:10.1128/JVI.80.1.20-26.2006. PMC 1317512. PMID 16352527.
- Crotty S, Cameron CE, Andino R (June 2001). "RNA virus error catastrophe: direct molecular test by using ribavirin". Proceedings of the National Academy of Sciences of the United States of America. 98 (12): 6895–900. Bibcode:2001PNAS...98.6895C. doi:10.1073/pnas.111085598. PMC 34449. PMID 11371613.
- Wilke CO (August 2005). "Quasispecies theory in the context of population genetics". BMC Evolutionary Biology. 5: 44. doi:10.1186/1471-2148-5-44. PMC 1208876. PMID 16107214.
- Wagner GP, Krall P (November 1993). "What is the difference between models of error thresholds and Muller's ratchet?". Journal of Mathematical Biology. 32 (1): 33–44. doi:10.1007/BF00160372.
- Sanjuán R, Moya A, Elena SF (June 2004). "The distribution of fitness effects caused by single-nucleotide substitutions in an RNA virus". Proceedings of the National Academy of Sciences of the United States of America. 101 (22): 8396–401. Bibcode:2004PNAS..101.8396S. doi:10.1073/pnas.0400146101. PMC 420405. PMID 15159545.
- Using fitness landscapes to visualize evolution in action, retrieved 2019-10-22
External links
- Video: Using fitness landscapes to visualize evolution in action—contains an example of "survival of the flattest"