2-Norbornyl cation
In organic chemistry, the term 2-norbornyl cation (or 2-bicyclo[2.2.1]heptyl cation) describes one of the three carbocations formed from derivatives of norbornane. Though 1-norbornyl and 7-norbornyl cations have been studied, the most extensive studies and vigorous debates have been centered on the exact structure of the 2-norbornyl cation.
The 2-norbornyl cation has been formed from a variety of norbornane derivatives and reagents. First reports of its formation and reactivity published by Saul Winstein sparked controversy over the nature of its bonding, as he invoked a three-center two-electron bond to explain the stereoselectivity of the resulting product. Herbert C. Brown challenged this assertion on the grounds that classical resonance structures could explain the stereospecificity without needing to adapt a new perspective of bonding. Both researchers' views had its supporters, and dozens of scientists contributed ingeniously designed experiments to provide evidence for one viewpoint or the other. Over time, the dispute became increasingly bitter and acrimonious, and the debate took on a personal or ad hominem character.[1]
Evidence of the non-classical nature of the 2-norbornyl cation grew over the course of several decades, mainly through spectroscopic data gathered using methods such as nuclear magnetic resonance (NMR). Crystallographic confirmation of its non-classical nature did not come until 2013.[2] Although most chemists now agree that 2-norbornyl cation itself is non-classical, it is also widely recognized that the energetic landscape for carbocations tends to be "flat", with many potential structures differing only minutely in energy. Certainly, not all bicyclic carbocations are non-classical; the energy difference between classical and non-classical structures is often delicately balanced. Thus, certain alkyl-substituted 2-bicyclo[2.2.1]heptyl cations are now known to adopt classical structures.
The nature of bonding in the 2-norbornyl cation incorporated many new ideas into the field’s understanding of chemical bonds. Similarities can be seen between this cation and others, such as boranes.
Theory
The nature of bonding in the 2-norbornyl cation was the center of a vigorous, well-known debate in the chemistry community through the middle of the twentieth century. While the majority of chemists believed that a three-center two-electron bond best depicted its ground state electronic structure, others argued that all data concerning the 2-norbornyl cation could be explained by depicting it as a rapidly equilibrating pair of cations.
At the height of the debate, all chemists agreed that the delocalized picture of electron bonding could be applied to the 2-norbornyl cation. But this did not answer the fundamental question on which the debate hinged. Researchers continued to search for novel ways to determine whether the three-centered delocalized picture described a low-energy transition state (saddle point on the multidimensional potential energy surface) or a potential energy minimum in its own right.[1] Proponents of the "classical" picture believed that the system was best described by a double-well potential with a very low barrier, while those in the "non-classical" camp envisioned the delocalized electronic state to describe a single potential energy well.[3]
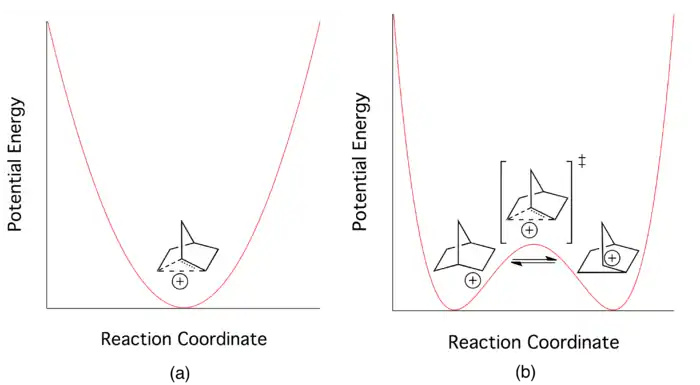
Hypovalency: the non-classical picture
Advocates of the non-classical nature of the stable 2-norbornyl cation typically depict the species using either resonance structures or a single structure with partial bonds (see Figure 2). This hypovalent interaction can be imagined as the net effect of i) a partial sigma bond between carbons 1 and 6, ii) a partial sigma bond between carbons 2 and 6, and iii) a partial pi bond between carbons 1 and 2.[4] Each partial bond is represented as a full bond in one of the three resonance structures or as a dashed partial bond if the cation is depicted through a single structure.

There has been some debate over how much the pi-bonded resonance structure actually contributes to the delocalized electronic structure.[6] Through 1H and 13C NMR spectroscopy, it has been confirmed that little positive charge lies on methylene carbon 6.[7] This is unsurprising as primary carbocations are much less stable than secondary carbocations. However, the 2-norbornyl cation can be formed from derivatives of β-(Δ3-cyclopentenyl)-ethane, indicating that the pi-bonded resonance structure is significant.[8]
The 2-norbornyl cation was one of the first examples of a non-classical ion. Non-classical ions can be defined as organic cations in which electron density of a filled bonding orbital is shared over three or more centers and contains some sigma-bond character.[9] The 2-norbornyl cation is seen as the prototype for non-classical ions. Other simple cations such as protonated acetylene (ethynium, C
2H+
3), protonated ethylene (ethenium, C
2H+
5), and protonated ethane (ethanium, C
2H+
7) have been shown to be best described as non-classical through infrared spectroscopy.[10]
The most frequently proposed molecular orbital depiction of the 2-norbornyl cation is shown in Figure 3. Two p-type orbitals, one on each of carbons 1 and 2, interact with a sp3-hybridized orbital on carbon 6 to form the hypovalent bond.[5][11] Extended Hückel Theory calculations for the 2-norbornyl cation suggest that the orbital on carbon 6 could instead be sp2-hybridized, though this only affects the geometry of the geminal hydrogens.[12]
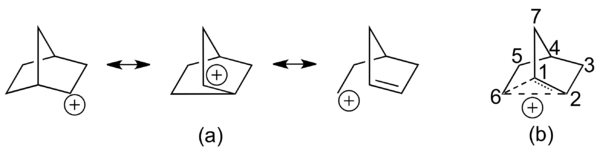
Rapid equilibrium: the classical picture

According to proponents of a classical double-well potential, the 2-norbornyl cation exists in dynamic equilibrium between two enantiomeric asymmetric structures. The delocalized species central to the non-classical picture is merely a transition state between the two structures. Wagner-Meerwein rearrangements are invoked as the mechanism that converts between the two enantiomers (see Figure 4).
Efforts to isolate the asymmetric species spectroscopically are typically unsuccessful. The major reason for this failure is reported to be extremely rapid forward and reverse reaction rates, which indicate a very low potential barrier for interconversion between the two enantiomers.[4]
Nortricyclonium: another non-classical structure
Some chemists have also considered the 2-norbornyl cation to be best represented by the nortricylconium ion, a C3-symmetric protonated nortricyclene. This depiction was first invoked to partially explain results of a 14C isotope scrambling experiment.[13] The molecular orbital representation of this structure involves an in-phase interaction between sp2-hybridized orbitals from carbons 1, 2 and 6 and the 1s atomic orbital on a shared hydrogen atom (see Figure 5).[14]
History
Non-classical ions
Non-classical ions differ from traditional cations in their electronic structure: though chemical bonds are typically depicted as the sharing of electrons between two atoms, stable non-classical ions can contain three or more atoms that share a single pair of electrons.[9] In 1939, Thomas Nevell and others attempted to elucidate the mechanism for transforming camphene hydrochloride into isobornyl chloride. In one of the proposed reaction mechanisms depicted in the paper, the positive charge of an intermediate cation was not assigned to a single atom but rather to the structure as a whole.[15] This was later cited by opponents of the non-classical description as the first time that a non-classical ion was invoked.[16] However, the term "non-classical ion" did not explicitly appear in the chemistry literature until over a decade later, when it was used to label delocalized bonding in a pyramidal, butyl cation.[17]
The term synartetic ion was also invoked to describe delocalized bonding in stable carbocations before the term non-classical ion was in widespread use. The first users of this term commented on the striking similarity between bonding in these types of cations and bonding in borohydrides.[18]
First non-classical proposals
In 1949, Saul Winstein observed that 2-exo-norbornyl brosylate (p-bromobenzenesulfonate) and 2-endo-norbornyl tosylate (p-toluenesulfonate) gave a racemic mixture of the same product, 2-exo-norbornyl acetate, upon acetolysis (see Figure 6). Since tosylates and brosylates work equally well as leaving groups, he concluded that both the 2-endo and 2-exo substituted norbornane must be going through a common cationic intermediate with a dominant exo reactivity. He reported that this intermediate was most likely a symmetric, delocalized 2-norbornyl cation.[19] It was later shown via vapor phase chromatography that the amount of the endo epimer of product produced was less than 0.02%, proving the high stereoselectivity of the reaction.[20]
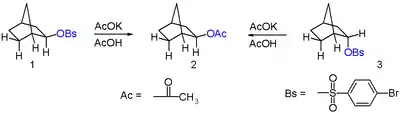
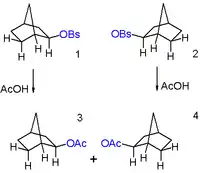
When a single enantiomer of 2-exo-norbornyl brosylate undergoes acetolysis, no optical activity is seen in the resulting 2-exo-norbornyl acetate (see Figure 7).[19] Under the non-classical description of the 2-norbornyl cation, the plane of symmetry present (running through carbons 4, 5, and 6) allow equal access to both enantiomers of the product, resulting in the observed racemic mixture.
It was also observed that the 2-exo-substituted norbornanes reacted 350 times faster than the corresponding endo isomers. Anchimeric assistance of the sigma bond between carbons 1 and 6 was rationalized as the explanation for this kinetic effect.[19] Importantly, the invoked anchimeric assistance led many chemists to postulate that the energetic stability of the 2-norbornyl cation was directly due to the symmetric, bridged structure invoked in the non-classical explanation. However, some other authors offered alternative explanations for the high stability without invoking a non-classical structure.[1]
In 1951, it was first suggested that the 2-norbornyl cation could actually be better described when viewed as a nortricyclonium ion.[13] It has been shown that the major product formed from an elimination reaction of the 2-norbornyl cation is nortricyclene (not norbornene), but this has been claimed to support both non-classical ion postulates.[20]
Herbert C. Brown: a dissenting view
Herbert C. Brown did not believe that it was necessary to invoke a new type of bonding in stable intermediates to explain the interesting reactivity of the 2-norbornyl cation. Criticizing many chemists for disregarding past explanations of reactivity, Brown argued that all of the aforementioned information about the 2-norbornyl cation could be explained using simple steric effects present in the norbornyl system.[4] Given that an alternative explanation using a rapidly equilibrating pair of ions for describing the 2-norbornyl cation was valid, he saw no need to invoke a stable, non-classical depiction of bonding.[21] Invoking stable non-classical ions was becoming commonplace; Brown felt that this was not only unwarranted but also counterproductive for the field of chemistry as a whole. Indeed, many papers reporting stable non-classical ions were later retracted for being unrealistic or incorrect.[3] After publishing this controversial view in 1962, Brown began a quest to find experimental evidence incompatible with the delocalized picture of bonding in the 2-norbornyl cation.[22]
Brown also worked to prove the instability of a delocalized electronic structure for the 2-norbornyl cation. If the non-classical ion could be proven to be higher in energy than the corresponding classical ion pair, the non-classical ion would only be seen as a transition state between the two asymmetric cations.[3][23] Though he did not rule out the possibility of a delocalized transition state Brown continued to reject the proposed reflectional symmetry of the 2-norbornyl cation, even late in his career.[24]
Impact
The introduction of the three-centered two-electron delocalized bond invoked in the non-classical picture of the 2-norbornyl cation allowed chemists to explore a whole new realm of chemical bonds. Chemists were eager to apply the characteristics of hypovalent electronic states to new and old systems alike (though several got too carried away).[6] One of the most fundamentally important concepts that emerged from the intense research focused around non-classical ions was the idea that electrons already involved in sigma bonds could be involved with reactivity. Though filled pi orbitals were known to be electron donors, chemists had doubted that sigma orbitals could function in the same capacity. The non-classical description of the 2-norbornyl cation can be seen as the donation of an electron pair from a carbon-carbon sigma bond into an empty p-orbital of carbon 2. Thus this carbocation showed that sigma-bond electron donation is as plausible as pi-bond electron donation.[25][26]
The intense debate that followed Brown’s challenge to non-classical ion proponents also had a large impact on the field of chemistry. In order to prove or disprove the non-classical nature of the 2-norbornyl cation, chemists on both sides of the debate zealously sought out new techniques for chemical characterization and more innovative interpretations of existing data.[27] One spectroscopic technique that was further developed to investigate the 2-norbornyl cation was nuclear magnetic resonance spectroscopy of compounds in highly acidic media.[1] Comparisons of the 2-norbornyl cation to unstable transition states with delocalized electronic states were often made when trying to elucidate whether the norbornyl system was stable or not. These efforts motivated closer investigations of transition states and vastly increased the scientific community’s understanding of their electronic structure.[27] In short, vigorous competition between scientific groups led to an extensive research and a better understanding of the underlying chemical concepts.
Formation
The 2-norbornyl cation can be made by a multitude of synthetic routes. These routes can be grouped into three different classes: σ Formation, π Formation, and Formation by Rearrangement. Each of these is discussed separately below.

σ formation
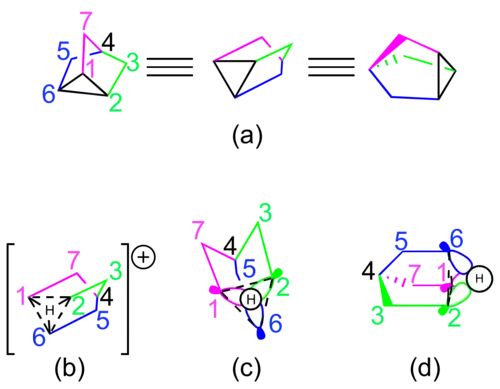
The starting material for this route is a norbornane derivative with a good leaving group in position 2. If the leaving group is on the exo- face, electron density from the σ bond between carbons 1 and 6 is donated into the σ* antibond between carbon 2 and the leaving group (see Figure 8b).[28]
If the leaving group is on the endo- face, the leaving group first leaves on its own. Then electron density from the σ bond between carbons 1 and 6 is donated into the resulting empty atomic orbital on carbon 2. However, this formation route is much slower than that of the exo- isomer because the σ bond cannot provide anchimeric assistance for the first step, making the activation energy to the first transition state much higher. Additionally, if there is a high concentration of reactive electrophiles in the reaction mixture, formation of a newly substituted norbornane derivative may preclude non-classical ion formation.[19][29]
An example of this formation route is the reaction that led Winstein and Trifan to propose the delocalized structure of the 2-norbornyl cation. 2-norbornyl tosylates and brosylates form the 2-norbornyl cation through this route as an intermediate towards solvolysis.[19]
π formation
The starting material for this route is a β-(Δ3-cyclopentenyl)-ethane derivative with a good leaving group on the terminal carbon of the ethane group. Electron density from the π bond of the alkene moiety is donated into the σ* anti-bond between the terminal carbon and the leaving group (see Figure 8c).[28][29]
For example, the major product of the acetolysis of β-(Δ3-cyclopentenyl)-ethyl nosylate (p-nitrobenzenesulfonate) is 2-exo-norbornyl acetate. The dearth of β-(Δ3-cyclopentenyl)-ethyl acetate present after the reaction is explained by the greater stability of the norbornyl system over the decorated cyclopentenyl system.[8]
This route is only effective if the cyclopentenyl olefin is isolated from any larger π-bonded system. The reaction rate significantly decreases if the involved double bond forms a six-membered aromatic ring as it does in 2-indanylethyl nosylate. Alkyl substitutions on the olefins have been seen to increase the reaction rate by stabilizing the resulting carbocation.[30]
Formation from rearrangement of 1-norbornyl and 7-norbornyl cations
The 2-norbornyl cation can also be formed via rearrangements of similar ions, such as the 1-norbornyl and 7-norbornyl cations, though these are generally not as well understood. Carbon-14 radioactive isotope labeling experiments have shown that complex scrambling in norbornyl cation systems allow 14C to be present at all seven positions of the norbornyl system.[13] By cycling between low and high temperatures during the hydrolyses of 1- and 7-choloronorbornanes, a large amount of 2-norbornanol was observed in addition to the expected 1- and 7-norbornanols, respectively. Thus the 1- and 7-norbornyl cations have some mechanism by which they can rearrange to the more stable 2-norbornyl cation on the timescale of solvolysis reactions.[31]
Geometry
Spectroscopic evidence
One probe for testing whether or not the 2-norbornyl cation is non-classical is investigating the inherent symmetry of the cation. Many spectroscopic tools, such as nuclear magnetic resonance spectroscopy (NMR spectroscopy) and Raman spectroscopy, give hints about the reflectional and rotational symmetry present in a molecule or ion. Each of the three proposed structures of the 2-norbornyl cation illustrates a different molecular symmetry. The non-classical form contains a reflection plane through carbons 4, 5, 6, and the midpoint of carbons 1 and 2. The classical form contains neither reflectional nor rotational symmetry. The protonated nortricyclene structure contains a C3-symmetric rotation axis through carbon 4.
Each peak in an NMR spectrum corresponds to a set of a particular element's atoms that are in similar chemical environments. The NMR spectrum of the antimony chloropentafluoride salt of the 2-norbornyl cation is not helpful at room temperature because hydride shifts occur faster than the timescale of an NMR experiment; most of the hydrogens are thus seen as equivalent and are accounted for in the same absorption peak. By lowering the temperature of the NMR experiment to −60 °C, hydride shifts are "frozen out" and more structural information can be gleaned from the spectrum. Researchers found that at these low temperatures, the 1H NMR spectrum matched what would be expected for the non-classical structure of the ion.[31][32]
1H and 13C NMR studies were able to confirm that any proposed Wagner-Meerwein rearrangements occurred faster than the timescale of the NMR experiment, even at low temperatures.[33] For molecules in static equilibrium with respect to rearrangements, NMR reveals how many sets of symmetry-related nuclei are in the molecule and how many nuclei each of these sets accounts for via spectrum integration. For molecules in dynamic equilibrium such as the 2-norbornyl cation, nuclei within each set can also be transformed to one another through rearrangements with fast reaction rates.[34] Since the proposed dynamic equilibrium of the classical ion proponents had very fast rates of rearrangement, the first NMR studies did not favor nor invalidate any of the three proposed structures.[7] But by using solid-state NMR analysis, one can lower the temperature of the NMR experiment to 5 kelvins (−268 °C) and thus significantly slow down any rearrangement phenomena. Solid-state 13C NMR spectra of the 2-norbornyl cation shows that carbons 1 and 2 are in identical chemical environments, which is consistent only with the non-classical picture of the 2-norbornyl cation.[35]
Raman spectra of the 2-norbornyl cation show a more symmetric species than would be expected for a pair of rapidly equilibrating classical ions. Since the proposed reaction rates for the classical ion rearrangements are slower than the Raman timescale, one would expect the Raman spectra to indicate a less symmetric species if the classical picture were correct.[6]
Some studies of the 13C NMR in particular favored interpretation via the protonated nortricyclene structure.[36] In addition, Raman spectra of the 2-norbornyl cation in some acidic solvents show an absorption band at 3110 cm-1 indicative of an electron-depleted cyclopropane ring. Since that absorption band would be expected in the C3-symmetric protonated nortricyclene, some scientists claimed this as convincing evidence for this interpretation.[37] Other chemists have postulated that the properties of the 2-norbornyl cation are very dependent on the solvent environment. Though the high acidity and low nucleophilicity of the solvents used in aforementioned experiments may cause the protonated nortricylconium geometry to be the most stable, this geometry need not be the most energetically favorable in other solvents.[6]
Calculations
Many calculational studies have been used to compare the feasibility of different proposed geometries. Using the quantum semi-empirical method of MINDO/3, researchers were not able to conclude which geometry of the 2-norbornyl cation was most energetically favorable. However, the classical structure was found to be the only potential minimum for the alkyl-substituted 2-methyl-2-norbornyl cation.[38] Additional calculations using Extended Hückel Theory for Molecular Orbitals were found to favor the non-classical geometry of the cation with reflectional symmetry.[12][39]
Thermodynamics
Some studies have used interesting comparisons in order to probe the energetic stability of the 2-norbornyl cation provided by its delocalized nature. Comparing the rearrangement between the 3-methyl-2-norbornyl cation and the 2-methyl-2-norbornyl cation to that between the tertiary and secondary isopentane carbocations, one finds that the change in enthalpy is about 6 kcal/mol less for the norbornyl system. Since the major difference between these two reversible rearrangements is the amount of delocalization possible in the electronic ground state, one can attribute the stabilization of the 3-methyl-2-norbornyl cation to its non-classical nature.[40] However, some experimental studies failed to observe this stabilization in solvolysis reactions.[3]
Other studies on the stability of the 2-norbornyl cation have shown that the alkyl substitutions at carbon 1 or 2 force the system to be decidedly classical. Tertiary carbocations are much more stable than their secondary counterparts and therefore do not need to adopt delocalized bonding in order to reach the lowest possible potential energy.[41][42]
Kinetics
To back up their suggestion of the non-classical nature of the 2-norbornyl cation, Winstein and Trifan first used kinetic evidence of the increased reaction rate for formation of the 2-exo-norbornyl cation over the 2-endo-norbornyl cation.[19] Other researchers investigated the reaction rate of compounds that could feature anchimeric assistance but could not undergo rearrangements as the norbornyl system could show similar trends in rate enhancement. This has been claimed by some to be definitive evidence for the non-classical picture.[43] But not all agree. Other researchers found that cyclopentane derivatives that were structurally similar to the norbornyl system still featured enhanced reaction rates, leading them to claim that the classical norbornyl cation describes the system much better.[44][45]
Isotope labeling experiments
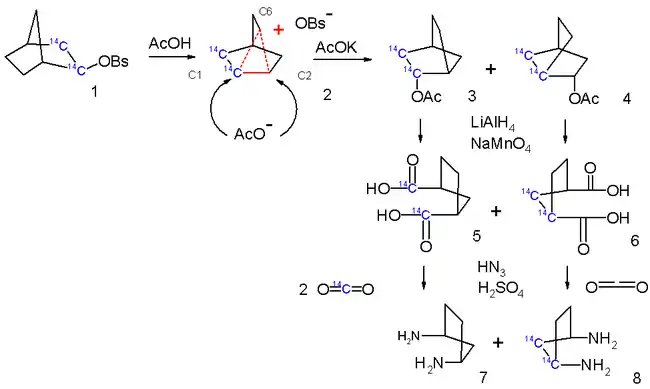
Radioactive isotope labeling experiments provide a powerful tool for determining the structure of organic molecules. By systematically decomposing the 2-norbornyl cation and analyzing the amount of radioactive isotope in each decomposition product, researchers were able to show further evidence for the non-classical picture of delocalized bonding (see Figure 9). Proponents of the nonclassical picture would expect 50% of the generated CO2 in the decomposition in Figure 9 to contain 14C, while proponents of the classical picture would expect more of the generated CO2 to be radioactive due to the short-lived nature of the cation. 40% of the carbon dioxide produced via decomposition has been observed to be radioactive, suggesting that the non-classical picture is more correct.[14]
Further distinction between non-classical and classical structures of the 2-norbornyl cation is possible by combining NMR experiments with isotope-labeling experiments. Isotopic substitution of one of two deuterium atoms for a hydrogen atom causes the environment of nearby NMR-active atoms to change dramatically. Asymmetric deuterium isotope labeling (substitution) will cause a set of carbons that were all equivalent in the all-hydrogen species to be split into two or more sets of equivalent carbons in the deutero-labeled species; this will be manifested in the NMR spectrum as one peak in the all-hydrogen species' spectrum becoming at least two "split" peaks in the deutero-labeled species. If a system is undergoing a rapid equilibrium at a rate faster than the timescale of a 13C NMR experiment, the relevant peak will be split dramatically (on the order of 10-100 ppm). If the system is instead static, the peak will be split very little.[46][47] The 13C NMR spectrum of the 2-norbornyl cation at -150 °C shows that the peaks corresponding to carbons 1 and 2 are split by less than 10 ppm (parts per million) when this experiment is carried out, indicating that the system is not undergoing a rapid equilibrium as in the classical picture.[48]
X-ray crystallography
Though characterization of 2-norbornyl cation crystals may have significantly precluded further debates about its electronic structure, it does not crystallize under any standard conditions. Recently, the crystal structure has been obtained and reported through a creative means: addition of aluminum tribromide to 2-norbornyl bromide in dibromomethane at low temperatures afforded crystals of [C
7H
11]+[Al
2Br
7]−·CH
2Br
2.[2] By examining the resulting crystal structure, researchers were able to confirm that the crystalline geometry best supports the case for delocalized bonding in the stable 2-norbornyl cation. Bond lengths between the "bridging" carbon 6 and each of carbons 1 and 2 were found to be slightly longer than typical alkane bonds. According to the nonclassical picture, one would expect a bond order between 0 and 1 for these bonds, signifying that this explains the crystal structure well. The bond length between carbons 1 and 2 was reported as being between typical single and double carbon-carbon bond lengths, which agrees with nonclassical predictions of a bond order slightly above 1. According to the non-classical picture, one would expect a bond order between 0 and 1 for the first two bonds. Investigators who crystallized the 2-norbornyl cation commented that the cation proved impossible to crystallize unless provided a chemical environment that locked it into one definite orientation.[2]
See also
References
- Walling, C. (1983). "An innocent bystander looks at the 2-norbornyl cation". Accounts of Chemical Research. 16 (12): 448. doi:10.1021/ar00096a004.
- Scholz, F.; Himmel, D.; Heinemann, F. W.; Schleyer, P. V. R.; Meyer, K.; Krossing, I. (2013). "Crystal Structure Determination of the Nonclassical 2-Norbornyl Cation". Science. 341 (6141): 62–64. Bibcode:2013Sci...341...62S. doi:10.1126/science.1238849. PMID 23828938.
- Brown, H. C. (1983). "The energy of the transition states and the intermediate cation in the ionization of 2-norbornyl derivatives. Where is the nonclassical stabilization energy?". Accounts of Chemical Research. 16 (12): 432. doi:10.1021/ar00096a002.
- Brown, H. C. (1973). "Question of σ Bridging in the solvolysis of 2-norbornyl derivatives". Accounts of Chemical Research. 6 (11): 377. doi:10.1021/ar50071a003.
- Streitwieser, Andrew (1961). "Chapter 12: Carbonium Ions". Molecular Orbital Theory for Organic Chemists. New York: John Wiley and Sons Inc. pp. 357–391.
- Sargent, George Dann (1971). "Chapter 24: The 2-Norbornyl Cation". In Olah, George; von Schleyer, Paul (eds.). Carbonium Ions. Volume III: Major Types (Continued). New York: Wiley-Interscience. pp. 1099–1200.
- Olah, G. A.; Liang, G.; Mateescu, G. D.; Riemenschneider, J. L. (1973). "Stable carbocations. CL. Fourier transform carbon-13 nuclear magnetic resonance and x-ray photoelectron spectroscopic study of the 2-norbornyl cation". Journal of the American Chemical Society. 95 (26): 8698. doi:10.1021/ja00807a032.
- Lawton, R. G. (1961). "1,5-Participation in the solvolysis of β-(Δ3-cyclopentenyl)ethyl p-nitrobenzenesulfonate". Journal of the American Chemical Society. 83 (10): 2399. doi:10.1021/ja01471a047.
- Sargent, G. D. (1966). "Bridged, non-classical carbonium ions". Quarterly Reviews, Chemical Society. 20 (2): 301–1073. doi:10.1039/QR9662000301.
-
Yeh, L. I.; Price, J. M.; Lee, Y. T. (1989). "Infrared spectroscopy of the pentacoordinated carbonium ion C
2H+
7". Journal of the American Chemical Society. 111 (15): 5597. doi:10.1021/ja00197a015. - Olah, G. A.; Mateescu, G. D.; Riemenschneider, J. L. (1972). "Electron spectroscopy of organic ions. II. Carbon 1s electron binding energies of the norbornyl, 2-methylnorbornyl, and related cations. Differentiation between nonclassical carbonium and classical carbenium ions". Journal of the American Chemical Society. 94 (7): 2529. doi:10.1021/ja00762a066.
- Trahanovsky, W. S. (1965). "Molecular Orbital Calculations of the Norbornyl Cation Using an Extended Hückel Theory". The Journal of Organic Chemistry. 30 (5): 1666. doi:10.1021/jo01016a517.
- J. D. Roberts and C. C. Lee (1951). "The nature of the intermediate in the solvolysis of norbornyl derivatives". J. Am. Chem. Soc. 73 (10): 5009–5010. doi:10.1021/ja01154a555.
- Roberts, J. D.; Lee, C. C.; Saunders, W. H. (1954). "Rearrangements in Carbonium Ion-Type Reactions of C14-Labeled Norbornyl Derivatives". Journal of the American Chemical Society. 76 (18): 4501. doi:10.1021/ja01647a001.
- Nevell, T. P.; De Salas, E.; Wilson, C. L. (1939). "259. Use of isotopes in chemical reactions. Part I. The mechanism of the Wagner–Meerwein rearrangement. Exchange of radioactive chlorine and of deuterium between camphene hydrochloride and hydrogen chloride". Journal of the Chemical Society (Resumed): 1188. doi:10.1039/JR9390001188.
- Brown, H. C.; Liu, K. T. (1975). "Additions to bicyclic olefins. VII. Electrophilic additions of hydrogen chloride and deuterium chloride to norbornene, 2-methylenenorbornane, and related bicyclic olefins. Evidence for a carbonium ion process and the capture of unsymmetrical (classical) 2-norbornyl cations". Journal of the American Chemical Society. 97 (3): 600. doi:10.1021/ja00836a022.
- Roberts, J. D.; Mazur, R. H. (1951). "The Nature of the Intermediate in Carbonium Ion-Type Interconversion Reactions of Cyclobutyl, Cyclopropylcarbinyl and Allylcarbinyl Derivatives". Journal of the American Chemical Society. 73 (7): 3542. doi:10.1021/ja01151a550.
- Brown, F.; Hughes, E. D.; Ingold, C. K.; Smith, J. F. (1951). "Wagner Changes, Synartetic Acceleration and Synartetic Ions". Nature. 168 (4263): 65. Bibcode:1951Natur.168...65B. doi:10.1038/168065a0.
- Winstein, S.; Trifan, D. S. (1949). "The Structure of the Bicyclo[2,2,1]2-Heptyl (Norbornyl) Carbonium Ion". Journal of the American Chemical Society. 71 (8): 2953. doi:10.1021/ja01176a536.
- Winstein, S.; Clippinger, E.; Howe, R.; Vogelfanger, E. (1965). "The Nonclassical Norbornyl Cation". Journal of the American Chemical Society. 87 (2): 376. doi:10.1021/ja01080a040.
- Brown, Herbert (1962). "Strained Transition States". Special Publication. No. 16: The Transition State. London: The Chemical Society. pp. 140–178.
- Brown, H. C.; Chloupek, F. J.; Rei, M. H. (1964). "Synthesis and Rates of Ethanolysis of 2-Phenyl-exo-norbornyl Chloride. The Question of a Nonclassical 1- and 2-Phenylnorbornyl Cation". Journal of the American Chemical Society. 86 (6): 1246. doi:10.1021/ja01060a058.
- Brown, H. C.; Morgan, K. J.; Chloupek, F. J. (1965). "Structural Effects in Solvolytic Reactions. I. The Role of Equilibrating Cations in Carbonium Ion Chemistry. Nature of the Intermediates Involved in the Solvolysis of Symmetrically Substituted β-Phenylethyl Derivatives". Journal of the American Chemical Society. 87 (10): 2137. doi:10.1021/ja01088a011.
- Brown, H. C. (1986). "Correspondence – the 2-norbornyl cation revisited". Accounts of Chemical Research. 19 (2): 34. doi:10.1021/ar00122a001.
- Olah, G. A. (1976). "Stable carbocations, 189. The σ-bridged 2-norbornyl cation and its significance to chemistry". Accounts of Chemical Research. 9 (2): 41. doi:10.1021/ar50098a001.
- Olah, G. A.; Prakash, G. K. S.; Saunders, M. (1983). "Conclusion of the classical-nonclassical ion controversy based on the structural study of the 2-norbornyl cation". Accounts of Chemical Research. 16 (12): 440. doi:10.1021/ar00096a003.
- Olah, G. A. (1995). "My Search for Carbocations and Their Role in Chemistry(Nobel Lecture)". Angewandte Chemie International Edition in English. 34 (1314): 1393–1405. doi:10.1002/anie.199513931.
- Winstein, S.; Carter, P. (1961). "The π-Route to a Bicycloöctyl Non-Classical Cation". Journal of the American Chemical Society. 83 (21): 4485. doi:10.1021/ja01482a057.
- Nenitzescu, Costin (1968). "Chapter 1: Historical Outlook". In Olah, George; von Schleyer, Paul (eds.). Carbonium Ions. Volume I: General Aspects and Methods of Investigation. New York: Wiley-Interscience. pp. 55–59.
- Bartlett, P. D.; Sargent, G. D. (1965). "Nucleophilic Reactivity of the Carbon–Carbon Double Bond. II. Solvolytic Ring Closure of 2-(3-methyl- and 3,4-dimethyl-Δ3-cyclopentenyl)ethyl p-nitrobenzenesulfonates". Journal of the American Chemical Society. 87 (6): 1297. doi:10.1021/ja01084a026.
- Schleyer, P. V. R.; Watts, W. E.; Fort, R. C.; Comisarow, M. B.; Olah, G. A. (1964). "Stable Carbonium Ions. X. Direct Nuclear Magnetic Resonance Observation of the 2-Norbornyl Cation". Journal of the American Chemical Society. 86 (24): 5679. doi:10.1021/ja01078a056.
- Olah, G. A.; Prakash, G. K. S.; Arvanaghi, M.; Anet, F. A. L. (1982). "High-Field 1H and 13C NMR Spectroscopic Study of the 2-Norbornyl Cation". Journal of the American Chemical Society. 104 (25): 7105. doi:10.1021/ja00389a037.
- Leone, Ronald; Barborak, J.C.; von Schleyer, Paul (1973). "Chapter 33: Degenerate Carbonium Ions". In Olah, George; von Schleyer, Paul (eds.). Carbonium Ions. Volume IV: Major Types (Continued). New York: Wiley-Interscience. pp. 1911–1915.
- Fry, James; Karabatsos, Gerasimos (1970). "Chapter 14: Intramolecular Hydride Shifts in Carbonium Ions". In Olah, George; von Schleyer, Paul (eds.). Carbonium Ions. Volume II: Methods of Formation and Major Types. New York: Wiley-Interscience. pp. 535–553.
- Yannoni, C. S.; Macho, V.; Myhre, P. C. (1982). "Resolved 13C NMR spectra of carbonium ions at cryogenic temperatures. The norbornyl cation at 5 K". Journal of the American Chemical Society. 104 (25): 7380. doi:10.1021/ja00389a108.
- Olah, G. A.; White, A. M. (1969). "Stable carbonium ions. LXXXVI. Carbon-13 nuclear magnetic resonance spectrum of the stable nonclassical norbornyl cation. Incompatibility with the equilibrating classical ion conception and further proof for the protonated nortricyclene structure". Journal of the American Chemical Society. 91 (14): 3954. doi:10.1021/ja01042a053.
- Olah, G. A.; Commeyras, A.; Lui, C. Y. (1968). "Stable carbonium ions. LXXII. Raman and N.M.R. Spectroscopic study of the nortricyclonium ion [protonated tricyclo[2.2.1.02,6]heptane] and its relation to the 2-norbornyl [bicyclo[2.2.1]heptyl] cation. The nature of the stable long-lived norbornyl cation in strong acid solutions". Journal of the American Chemical Society. 90 (14): 3882. doi:10.1021/ja01016a062.
- Radom, Leo; Poppinger, Dieter; Haddon, Robert (1976). "Chapter 38: Molecular Orbital Theory of Carbocations". In Olah, George; von Schleyer, Paul (eds.). Carbonium Ions. Volume V: Miscellaneous Ions, Theory, and Structure. New York: Wiley-Interscience. pp. 2390–2391.
- Hoffmann, R. (1964). "Extended Hückel Theory. IV. Carbonium Ions". The Journal of Chemical Physics. 40 (9): 2480. Bibcode:1964JChPh..40.2480H. doi:10.1063/1.1725551.
- Schleyer, P. V. R.; Chandrasekhar, J. (1981). "Evaluation of the extra stability of the bridged 2-norbornyl cation". The Journal of Organic Chemistry. 46: 225. doi:10.1021/jo00314a065.
- Brown, H. C.; Rei, M. H. (1964). "Comparison of the Effect of Substituents at the 2-Position of the Norbornyl System with Their Effect in Representative Secondary Aliphatic and Alicyclic Derivatives. Evidence for the Absence of Nonclassical Stabilization of the Norbornyl Cation". Journal of the American Chemical Society. 86 (22): 5008. doi:10.1021/ja01076a058.
- Bunton, C.A. (1963). "Chapter 2. Structural Effects upon Rate of Substitution; Section 11. Non-classical carbonium ions". In Hughes, E.D. (ed.). Reaction Mechanisms in Organic Chemistry. Volume I: Nucleophilic Substitution at a Saturated Carbon Atom. Amsterdam: Elsevier Publishing Company. pp. 59–67.
- Schleyer, P. V. R. (1964). "The Nonclassical Carbonium Ion Problem: Reaction Rates". Journal of the American Chemical Society. 86 (9): 1856. doi:10.1021/ja01063a044.
- Brown, H. C.; Chloupek, F. J.; Rei, M. H. (1964). "Comparison of the Rates of Solvolysis of Representative Norbornyl and Cyclopentyl Derivatives. A Critical Examination of Rates as a Basis for the Postulated Nonclassical Structure of Norbornyl Cations". Journal of the American Chemical Society. 86 (6): 1247. doi:10.1021/ja01060a059.
- Brown, H. C.; Chloupek, F. J.; Rei, M. H. (1964). "Rates of Solvolysis of the p-Nitrobenzoates of exo-endo' Tertiary Norborneols. A Critical Examination of the exo-endo Rate Ratio as a Basis for the Postulated Nonclassical Structure of the Norbornyl Cation". Journal of the American Chemical Society. 86 (6): 1248. doi:10.1021/ja01060a060.
- Saunders, M.; Telkowski, L.; Kates, M. R. (1977). "Isotopic perturbation of degeneracy. Carbon-13 nuclear magnetic resonance spectra of dimethylcyclopentyl and dimethylnorbornyl cations". Journal of the American Chemical Society. 99 (24): 8070. doi:10.1021/ja00466a060.
- Saunders, M.; Kates, M. R. (1977). "Isotopic perturbation of resonance. Carbon-13 nuclear magnetic resonance spectra of deuterated cyclohexenyl and cyclopentenyl cations". Journal of the American Chemical Society. 99 (24): 8071. doi:10.1021/ja00466a061.
- Saunders, M.; Kates, M. R. (1980). "Deuterium isotope effect on the carbon-13 NMR spectrum of the bicyclo[2.2.1]heptyl cation. Nonclassical norbornyl cation". Journal of the American Chemical Society. 102 (22): 6867. doi:10.1021/ja00542a044.