Elimination reaction
An elimination reaction is a type of organic reaction in which two substituents are removed from a molecule in either a one- or two-step mechanism.[2] The one-step mechanism is known as the E2 reaction, and the two-step mechanism is known as the E1 reaction. The numbers refer not to the number of steps in the mechanism, but rather to the kinetics of the reaction: E2 is bimolecular (second-order) while E1 is unimolecular (first-order). In cases where the molecule is able to stabilize an anion but possesses a poor leaving group, a third type of reaction, E1CB, exists. Finally, the pyrolysis of xanthate and acetate esters proceed through an "internal" elimination mechanism, the Ei mechanism.
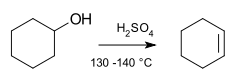
Loss of hydron (H+)
In most organic elimination reactions, at least one hydron (H+) is lost to form the double bond: the unsaturation of the molecule increases. It is also possible that a molecule undergoes reductive elimination, by which the valence of an atom in the molecule decreases by two, though this is more common in inorganic chemistry. An important class of elimination reactions is those involving alkyl halides, with good leaving groups, reacting with a Lewis base to form an alkene. Elimination may be considered the reverse of an addition reaction. When the substrate is asymmetric, regioselectivity is determined by Zaitsev's rule or through Hofmann elimination if the carbon with the most substituted hydrogen is inaccessible.
E2 mechanism
During the 1920s, Christopher Kelk Ingold proposed a model to explain a peculiar type of chemical reaction: the E2 mechanism. E2 stands for bimolecular elimination. The reaction involves a one-step mechanism in which carbon-hydrogen and carbon-halogen bonds break to form a double bond (C=C Pi bond).
The specifics of the reaction are as follows:
- E2 is a single step elimination, with a single transition state.
- It is typically undergone by primary substituted alkyl halides, but is possible with some secondary alkyl halides and other compounds.
- The reaction rate is second order, because it's influenced by both the alkyl halide and the base (bimolecular).
- Because the E2 mechanism results in the formation of a pi bond, the two leaving groups (often a hydrogen and a halogen) need to be antiperiplanar. An antiperiplanar transition state has staggered conformation with lower energy than a synperiplanar transition state which is in eclipsed conformation with higher energy. The reaction mechanism involving staggered conformation is more favorable for E2 reactions (unlike E1 reactions).
- E2 typically uses a strong base. It must be strong enough to remove a weakly acidic hydrogen.
- In order for the pi bond to be created, the hybridization of carbons needs to be lowered from sp3 to sp2.
- The C-H bond is weakened in the rate determining step and therefore a primary deuterium isotope effect much larger than 1 (commonly 2-6) is observed.
- E2 competes with the SN2 reaction mechanism if the base can also act as a nucleophile (true for many common bases).
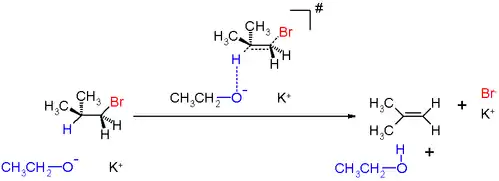
An example of this type of reaction in scheme 1 is the reaction of isobutylbromide with potassium ethoxide in ethanol. The reaction products are isobutene, ethanol and potassium bromide.
E1 mechanism
E1 is a model to explain a particular type of chemical elimination reaction. E1 stands for unimolecular elimination and has the following specifications
- It is a two-step process of elimination: ionization and deprotonation.
- Ionization: the carbon-halogen bond breaks to give a carbocation intermediate.
- deprotonation of the carbocation.
- E1 typically takes place with tertiary alkyl halides, but is possible with some secondary alkyl halides.
- The reaction rate is influenced only by the concentration of the alkyl halide because carbocation formation is the slowest step, as known as the rate-determining step. Therefore, first-order kinetics apply (unimolecular).
- The reaction usually occurs in the complete absence of a base or the presence of only a weak base (acidic conditions and high temperature).
- E1 reactions are in competition with SN1 reactions because they share a common carbocationic intermediate.
- A secondary deuterium isotope effect of slightly larger than 1 (commonly 1 - 1.5) is observed.
- There is no antiperiplanar requirement. An example is the pyrolysis of a certain sulfonate ester of menthol:

- Only reaction product A results from antiperiplanar elimination. The presence of product B is an indication that an E1 mechanism is occurring.[3]
- It is accompanied by carbocationic rearrangement reactions
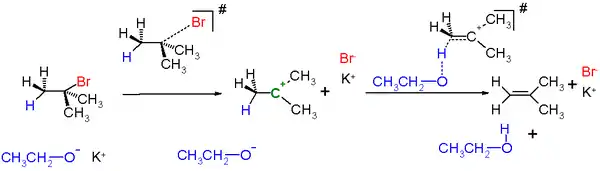
An example in scheme 2 is the reaction of tert-butylbromide with potassium ethoxide in ethanol.
E1 eliminations happen with highly substituted alkyl halides for two main reasons.
- Highly substituted alkyl halides are bulky, limiting the room for the E2 one-step mechanism; therefore, the two-step E1 mechanism is favored.
- Highly substituted carbocations are more stable than methyl or primary substituted cations. Such stability gives time for the two-step E1 mechanism to occur.
- If SN1 and E1 pathways are competing, the E1 pathway can be favored by increasing the heat.
Specific features :
- Rearrangement possible
- Independent of concentration and basicity of base
Competition among mechanisms
The reaction rate is influenced by the reactivity of halogens, iodide and bromide being favored. Fluoride is not a good leaving group, so eliminations with fluoride as the leaving group have slower rates than other halogens. There is a certain level of competition between the elimination reaction and nucleophilic substitution. More precisely, there are competitions between E2 and SN2 and also between E1 and SN1. Substitution generally predominates and elimination occurs only during precise circumstances. Generally, elimination is favored over substitution when
- steric hindrance around the α-carbon increases.
- a stronger base is used.
- temperature increases (increase entropy)
- the base is a poor nucleophile. Bases with steric bulk, (such as in Potassium tert-butoxide), are often poor nucleophiles.
In one study[4] the kinetic isotope effect (KIE) was determined for the gas phase reaction of several alkyl halides with the chlorate ion. In accordance with an E2 elimination the reaction with t-butyl chloride results in a KIE of 2.3. The methyl chloride reaction (only SN2 possible) on the other hand has a KIE of 0.85 consistent with a SN2 reaction because in this reaction type the C-H bonds tighten in the transition state. The KIE's for the ethyl (0.99) and isopropyl (1.72) analogues suggest competition between the two reaction modes.
Elimination reactions other than β-elimination
β-Elimination, with loss of electrofuge and nucleofuge on vicinal carbons, is by far the most common type of elimination. The ability to form a stable product containing a C=C or C=X bond, as well as orbital alignment considerations, strongly favors β-elimination over other elimination processes.[5] However, other types are known, generally for systems where β-elimination cannot occur.
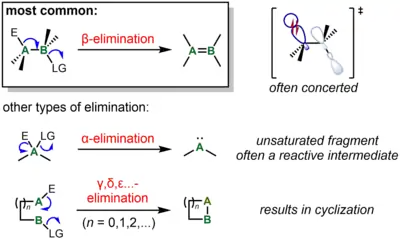
The next most common type of elimination reaction is α-elimination. For a carbon center, the result of α-elimination is the formation of a carbene, which includes "stable carbenes" such as carbon monoxide or isocyanides. For instance, α-elimination the elements of HCl from chloroform (CHCl3) in the presence of strong base is a classic approach for the generation of dichlorocarbene, :CCl2, as a reactive intermediate. On the other hand, formic acid undergoes α-elimination to afford the stable products water and carbon monoxide under acidic conditions. α-Elimination may also occur on a metal center, one particularly common result of which is lowering of both the metal oxidation state and coordination number by 2 units in a process known as reductive elimination. (Confusingly, in organometallic terminology, the terms α-elimination and α-abstraction refer to processes that result in formation of a metal-carbene complex.[6] In these reactions, it is the carbon adjacent to the metal that undergoes α-elimination.)
In certain special cases, γ- and higher eliminations to form three-membered or larger rings is also possible in both organic and organometallic processes. For instance, certain Pt(II) complexes undergo γ- and δ-elimination to give metallocycles.[7] More recently, γ-silyl elimination of a silylcyclobutyl tosylate has been used to prepare strained bicyclic systems.[8]
See also
References
- Coleman, G. H.; Johnstone, H. F. (1925). "Cyclohexene". Organic Syntheses. 5: 33. doi:10.15227/orgsyn.005.0033.
- March, Jerry (1985), Advanced Organic Chemistry: Reactions, Mechanisms, and Structure (3rd ed.), New York: Wiley, ISBN 0-471-85472-7
- Nash, J. J.; Leininger, M. A.; Keyes, K. (April 2008). "Pyrolysis of Aryl Sulfonate Esters in the Absence of Solvent: E1 or E2? A Puzzle for the Organic Laboratory". Journal of Chemical Education. 85 (4): 552. Bibcode:2008JChEd..85..552N. doi:10.1021/ed085p552.
- Stephanie M. Villano; Shuji Kato; Veronica M. Bierbaum (2006). "Deuterium Kinetic Isotope Effects in Gas-Phase SN2 and E2 Reactions: Comparison of Experiment and Theory". J. Am. Chem. Soc. 128 (3): 736–737. doi:10.1021/ja057491d. PMID 16417360.
- 1960-, Anslyn, Eric V. (2006). Modern physical organic chemistry. Dougherty, Dennis A., 1952-. Sausalito, CA: University Science. ISBN 1891389319. OCLC 55600610.CS1 maint: numeric names: authors list (link)
- 1948-, Crabtree, Robert H. (2009). The organometallic chemistry of the transition metals (5th ed.). Hoboken, N.J.: Wiley. ISBN 9780470257623. OCLC 268790870.CS1 maint: numeric names: authors list (link)
- Moore, Stephen S.; DiCosimo, Robert; Sowinski, Allan F.; Whitesides, George M. (1981-02-01). "Ring strain in bis(triethylphosphine)-3,3-dimethylplatinacyclobutane is small". Journal of the American Chemical Society. 103 (4): 948–949. doi:10.1021/ja00394a043. ISSN 0002-7863.
- Kelly, Christopher B.; Colthart, Allison M.; Constant, Brad D.; Corning, Sean R.; Dubois, Lily N. E.; Genovese, Jacqueline T.; Radziewicz, Julie L.; Sletten, Ellen M.; Whitaker, Katherine R. (2011-04-01). "Enabling the Synthesis of Perfluoroalkyl Bicyclobutanes via 1,3 γ-Silyl Elimination". Organic Letters. 13 (7): 1646–1649. doi:10.1021/ol200121f. ISSN 1523-7060. PMID 21366262.
External links
 Media related to Elimination reactions at Wikimedia Commons
Media related to Elimination reactions at Wikimedia Commons Quotations related to Elimination reaction at Wikiquote
Quotations related to Elimination reaction at Wikiquote
| Authority control |
|---|
