Pyruvate dehydrogenase complex
Pyruvate dehydrogenase complex (PDC) is a complex of three enzymes that converts pyruvate into acetyl-CoA by a process called pyruvate decarboxylation.[1] Acetyl-CoA may then be used in the citric acid cycle to carry out cellular respiration, and this complex links the glycolysis metabolic pathway to the citric acid cycle. Pyruvate decarboxylation is also known as the "pyruvate dehydrogenase reaction" because it also involves the oxidation of pyruvate.[2]
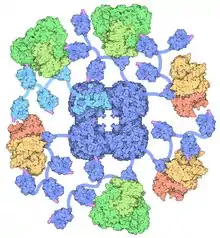
This multi-enzyme complex is related structurally and functionally to the oxoglutarate dehydrogenase and branched-chain oxo-acid dehydrogenase multi-enzyme complexes.
Reaction
The reaction catalysed by pyruvate dehydrogenase complex is:
| pyruvate | pyruvate dehydrogenase complex | acetyl CoA | |
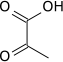 |
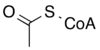 | ||
| CoA-SH + NAD+ | CO2 + NADH + H+ | ||
Structure
| Enzymes | Abbrev. | Cofactors | # subunits prokaryotes | # subunits eukaryotes |
|---|---|---|---|---|
| pyruvate dehydrogenase (EC 1.2.4.1) | E1 | TPP (thiamine pyrophosphate) | 24 | 30 |
| dihydrolipoyl transacetylase (EC 2.3.1.12) | E2 | lipoate coenzyme A | 24 | 60 |
| dihydrolipoyl dehydrogenase (EC 1.8.1.4) | E3 | FAD NAD+ | 12 | 12 |
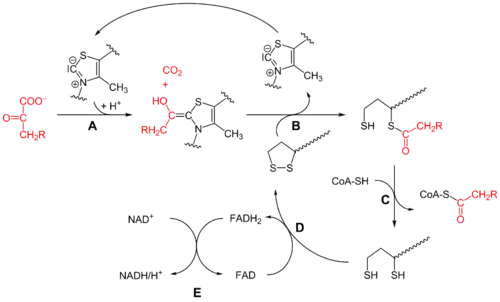
Pyruvate dehydrogenase (E1)
Initially, pyruvate and thiamine pyrophosphate (TPP or vitamin B1) are bound by pyruvate dehydrogenase subunits.[1] The thiazolium ring of TPP is in a zwitterionic form, and the anionic C2 carbon performs a nucleophilic attack on the C2 (ketone) carbonyl of pyruvate. The resulting hemithioacetal undergoes decarboxylation to produce an acyl anion equivalent (see cyanohydrin or aldehyde-dithiane umpolung chemistry, as well as benzoin condensation). This anion attacks S1 of an oxidized lipoate species that is attached to a lysine residue. In a ring-opening SN2-like mechanism, S2 is displaced as a sulfide or sulfhydryl moiety. Subsequent collapse of the tetrahedral hemithioacetal ejects thiazole, releasing the TPP cofactor and generating a thioacetate on S1 of lipoate. The E1-catalyzed process is the rate-limiting step of the whole pyruvate dehydrogenase complex.
Dihydrolipoyl transacetylase (E2)
At this point, the lipoate-thioester functionality is translocated into the dihydrolipoyl transacetylase (E2) active site,[1] where a transacylation reaction transfers the acetyl from the "swinging arm" of lipoyl to the thiol of coenzyme A. This produces acetyl-CoA, which is released from the enzyme complex and subsequently enters the citric acid cycle. E2 can also be known as lipoamide reductase-transacetylase.
Dihydrolipoyl dehydrogenase (E3)
The dihydrolipoate, still bound to a lysine residue of the complex, then migrates to the dihydrolipoyl dehydrogenase (E3) active site,[1] where it undergoes a flavin-mediated oxidation, identical in chemistry to disulfide isomerase. First, FAD oxidizes dihydrolipoate back to its lipoate resting state, producing FADH2. Then, a NAD+ cofactor oxidizes FADH2 back to its FAD resting state, producing NADH.
Structural differences between species
PDC is a large complex composed of multiple copies of 3 or 4 subunits depending on species.
Gram-negative bacteria
In Gram-negative bacteria, e.g. Escherichia coli, PDC consists of a central cubic core made up from 24 molecules of dihydrolipoyl transacetylase (E2). Up to 24 copies of pyruvate dehydrogenase (E1) and 12 molecules of dihydrolipoyl dehydrogenase (E3) bind to the outside of the E2 core.[3]
Gram-positive bacteria and eukaryotes
In contrast, in Gram-positive bacteria (e.g. Bacillus stearothermophilus) and eukaryotes the central PDC core contains 60 E2 molecules arranged into an icosahedron.
Eukaryotes also contain 12 copies of an additional core protein, E3 binding protein (E3BP). The exact location of E3BP is not completely clear. Cryo-electron microscopy has established that E3BP binds to each of the icosahedral faces in yeast.[4] However, it has been suggested that it replaces an equivalent number of E2 molecules in the bovine PDC core.
Up to 60 E1 or E3 molecules can associate with the E2 core from Gram-positive bacteria - binding is mutually exclusive. In eukaryotes E1 is specifically bound by E2, while E3 associates with E3BP. It is thought that up to 30 E1 and 6 E3 enzymes are present, although the exact number of molecules can vary in vivo and often reflects the metabolic requirements of the tissue in question.
Regulation
Pyruvate dehydrogenase is inhibited when one or more of the three following ratios are increased: ATP/ADP, NADH/NAD+ and acetyl-CoA/CoA.
In eukaryotes PDC is tightly regulated by its own specific pyruvate dehydrogenase kinase (PDK) and pyruvate dehydrogenase phosphatase (PDP), deactivating and activating it respectively.[5]
- PDK phosphorylates three specific serine residues on E1 with different affinities. Phosphorylation of any one of them (using ATP) renders E1 (and in consequence the entire complex) inactive.[5]
- Dephosphorylation of E1 by PDP reinstates complex activity.[5]
Products of the reaction act as allosteric inhibitors of the PDC, because they activate PDK. Substrates in turn inhibit PDK, reactivating PDC.
During starvation, PDK increases in amount in most tissues, including skeletal muscle, via increased gene transcription. Under the same conditions, the amount of PDP decreases. The resulting inhibition of PDC prevents muscle and other tissues from catabolizing glucose and gluconeogenesis precursors. Metabolism shifts toward fat utilization, while muscle protein breakdown to supply gluconeogenesis precursors is minimized, and available glucose is spared for use by the brain.
Calcium ions have a role in regulation of PDC in muscle tissue, because it activates PDP, stimulating glycolysis on its release into the cytosol - during muscle contraction. Some products of these transcriptions release H2 into the muscles. This can cause calcium ions to decay over time.
Localization of pyruvate decarboxylation
In eukaryotic cells the pyruvate decarboxylation occurs inside the mitochondrial matrix, after transport of the substrate, pyruvate, from the cytosol. The transport of pyruvate into the mitochondria is via the transport protein pyruvate translocase. Pyruvate translocase transports pyruvate in a symport fashion with a proton, and hence is active, consuming energy.. Alternative sources say "transport of pyruvate across the outer mitochondrial membrane appears to be easily accomplished via large non-selective channels such as voltage-dependent anion channels, which enable passive diffusion" and transport across inner mitochondrial membrane is mediated by mitochondrial pyruvate carrier 1 (MPC1) and mitochondrial pyruvate carrier 2 (MPC2).[6]
Upon entry to the mitochondria, the pyruvate is decarboxylated, producing acetyl-CoA. This irreversible reaction traps the acetyl-CoA within the mitochondria (the acetyl-CoA can only be transported out of the mitochondrial matrix under conditions of high oxaloacetate via the citrate shuttle, a TCA intermediate that is normally sparse). The carbon dioxide produced by this reaction is nonpolar and small, and can diffuse out of the mitochondria and out of the cell.
In prokaryotes, which have no mitochondria, this reaction is either carried out in the cytosol, or not at all.
Clinical relevance
Pyruvate dehydrogenase deficiency can result from mutations in any of the enzymes or cofactors. Its primary clinical finding is lactic acidosis.[7]
See also
References
- DeBrosse, Suzanne D.; Kerr, Douglas S. (2016-01-01), Saneto, Russell P.; Parikh, Sumit; Cohen, Bruce H. (eds.), "Chapter 12 - Pyruvate Dehydrogenase Complex Deficiency", Mitochondrial Case Studies, Boston: Academic Press, pp. 93–101, doi:10.1016/b978-0-12-800877-5.00012-7, ISBN 978-0-12-800877-5, retrieved 2020-11-16
- J. M. Berg; J. L. Tymoczko, L. Stryer (2007). Biochemistry (6 ed.). Freeman. ISBN 978-0-7167-8724-2.CS1 maint: multiple names: authors list (link)
- Izard T, Aevarsson A, Allen MD, Westphal AH, Perham RN, de Kok A, Hol WG (1999). "Principles of quasi-equivalence and Euclidean geometry govern the assembly of cubic and dodecahedral cores of pyruvate dehydrogenase complexes". Proc. Natl. Acad. Sci. USA. 96 (4): 1240–1245. Bibcode:1999PNAS...96.1240I. doi:10.1073/pnas.96.4.1240. PMC 15447. PMID 9990008.
- Stoops, J.K., Cheng, R.H., Yazdi, M.A., Maeng, C.Y., Schroeter, J.P., Klueppelberg, U., Kolodziej, S.J., Baker, T.S., Reed, L.J. (1997) On the unique structural organization of the Saccharomyces cerevisiae pyruvate dehydrogenase complex. J. Biol. Chem. 272, 5757-5764.
- Pelley, John W. (2012-01-01), Pelley, John W. (ed.), "6 - Glycolysis and Pyruvate Oxidation", Elsevier's Integrated Review Biochemistry (Second Edition), Philadelphia: W.B. Saunders, pp. 49–55, doi:10.1016/b978-0-323-07446-9.00006-4, ISBN 978-0-323-07446-9, retrieved 2020-11-16
- Rutter, Jared (23 January 2013). "The long and winding road to the mitochondrial pyruvate carrier". Cancer & Metabolism. doi:10.1186/2049-3002-1-6. PMID 24280073.
- "Pyruvate dehydrogenase deficiency". Genetics Home Reference. Retrieved March 17, 2013.
External links
| Wikimedia Commons has media related to Pyruvate dehydrogenase complex. |
- https://web.archive.org/web/20070405211049/http://www.dentistry.leeds.ac.uk/biochem/MBWeb/mb1/part2/krebs.htm#animat1 - animation of the general mechanism of the PDC (link on upper right) at University of Leeds
- Pyruvate+Dehydrogenase+Complex at the US National Library of Medicine Medical Subject Headings (MeSH)
3D structures
- Zhou, H.; McCarthy, B.; O'Connor, M.; Reed, J.; Stoops, K. (Dec 2001). "The remarkable structural and functional organization of the eukaryotic pyruvate dehydrogenase complexes". Proceedings of the National Academy of Sciences of the United States of America. 98 (26): 14802–14807. Bibcode:2001PNAS...9814802Z. doi:10.1073/pnas.011597698. ISSN 0027-8424. PMC 64939. PMID 11752427., bovine kidney pyruvate dehydrogenase complex
- Yu, X.; Hiromasa, Y.; Tsen, H.; Stoops, K.; Roche, E.; Zhou, H. (Jan 2008). "Structures of the Human Pyruvate Dehydrogenase Complex Cores: A Highly Conserved Catalytic Center with Flexible N-Terminal Domains". Structure. 16 (1): 104–114. doi:10.1016/j.str.2007.10.024. ISSN 0969-2126. PMC 4807695. PMID 18184588., human full-length and truncated E2 (tE2) cores of PDC, expressed in E. coli
