DNA oxidation
DNA oxidation is the process of oxidative damage of deoxyribonucleic acid. As described in detail by Burrows et al.,[1] 8-oxo-2'-deoxyguanosine (8-oxo-dG) is the most common oxidative lesion observed in duplex DNA because guanine has a lower one-electron reduction potential than the other nucleosides in DNA. The one electron reduction potentials of the nucleosides (in volts versus NHE) are guanine 1.29, adenine 1.42, cytosine 1.6 and thymine 1.7. About 1 in 40,000 guanines in the genome are present as 8-oxo-dG under normal conditions. This means that >30,000 8-oxo-dGs may exist at any given time in the genome of a human cell. Another product of DNA oxidation is 8-oxo-dA. 8-oxo-dA occurs at about 1/3 the frequency of 8-oxo-dG. The reduction potential of guanine may be reduced by as much as 50%, depending on the particular neighboring nucleosides stacked next to it within DNA.
Excess DNA oxidation is linked to certain diseases and cancers,[2] while normal levels of oxidized nucleotides, due to normal levels of ROS, may be necessary for memory and learning.[3][4]
Oxidized bases in DNA
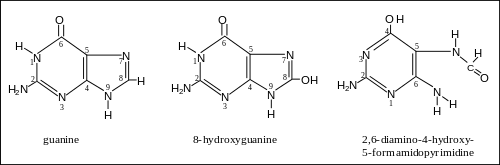
More than 20 oxidatively damaged DNA base lesions were identified in 2003 by Cooke et al.[5] and these overlap the 12 oxidized bases reported in 1992 by Dizdaroglu.[6] Two of the most frequently oxidized bases found by Dizdaroglu after ionizing radiation (causing oxidative stress) were the two oxidation products of guanine shown in the figure. One of these products was 8-OH-Gua (8-hydroxyguanine). (The article 8-oxo-2'-deoxyguanosine refers to the same damaged base since the keto form 8-oxo-Gua described there may undergo a tautomeric shift to the enol form 8-OH-Gua shown here.) The other product was FapyGua (2,6-diamino-4-hydroxy-5-formamidopyrimidine). Another frequent oxidation product was 5-OH-Hyd (5-hydroxyhydantoin) derived from cytosine.
Removal of oxidized bases
Most oxidized bases are removed from DNA by enzymes operating within the base excision repair pathway.[5] Removal of oxidized bases in DNA is fairly rapid. For example, 8-oxo-dG was increased 10-fold in the livers of mice subjected to ionizing radiation, but the excess 8-oxo-dG was removed with a half-life of 11 minutes.[7]
Steady-state levels of DNA damages
Steady-state levels of endogenous DNA damages represent the balance between formation and repair. Swenberg et al.[8] measured average amounts of steady state endogenous DNA damages in mammalian cells. The seven most common damages they found are shown in Table 1. Only one directly oxidized base, 8-hydroxyguanine, at about 2,400 8-OH-G per cell, was among the most frequent DNA damages present in the steady-state.
| Endogenous lesions | Number per cell |
|---|---|
| Abasic sites | 30,000 |
| N7-(2-hydroxethyl)guanine (7HEG) | 3,000 |
| 8-hydroxyguanine | 2,400 |
| 7-(2-oxoethyl)guanine | 1,500 |
| Formaldehyde adducts | 960 |
| Acrolein-deoxyguanine | 120 |
| Malondialdehyde-deoxyguanine | 60 |
Increased 8-oxo-dG in carcinogenesis and disease
_and_with_tumorigenesis_(B)._Brown_shows_8-oxo-dG.jpg.webp)
As reviewed by Valavanidis et al.[10] increased levels of 8-oxo-dG in a tissue can serve as a biomarker of oxidative stress. They also noted that increased levels of 8-oxo-dG are frequently found associated with carcinogenesis and disease.
In the figure shown in this section, the colonic epithelium from a mouse on a normal diet has a low level of 8-oxo-dG in its colonic crypts (panel A). However, a mouse likely undergoing colonic tumorigenesis (due to deoxycholate added to its diet[9]) has a high level of 8-oxo-dG in its colonic epithelium (panel B). Deoxycholate increases intracellular production of reactive oxygen resulting in increased oxidative stress,[11][12] and this may contribute to tumorigenesis and carcinogenesis. Of 22 mice fed the diet supplemented with deoxycholate, 20 (91%) developed colonic tumors after 10 months on the diet, and the tumors in 10 of these mice (45% of mice) included an adenocarcinoma (cancer).[9] Cooke et al.[5] point out that a number of diseases, such as Alzheimer's disease and systemic lupus erythematosus, have elevated 8-oxo-dG but no increased carcinogenesis.
Indirect role of oxidative damage in carcinogenesis
Valavanidis et al.[10] pointed out that oxidative DNA damage, such as 8-oxo-dG, may contribute to carcinogenesis by two mechanisms. The first mechanism involves modulation of gene expression, whereas the second is through the induction of mutations.
Epigenetic alterations
Epigenetic alteration, for instance by methylation of CpG islands in a promoter region of a gene, can repress expression of the gene (see DNA methylation in cancer). In general, epigenetic alteration can modulate gene expression. As reviewed by Bernstein and Bernstein,[13] the repair of various types of DNA damages can, with low frequency, leave remnants of the different repair processes and thereby cause epigenetic alterations. 8-oxo-dG is primarily repaired by base excision repair (BER).[14] Li et al.[15] reviewed studies indicating that one or more BER proteins also participate(s) in epigenetic alterations involving DNA methylation, demethylation or reactions coupled to histone modification. Nishida et al.[16] examined 8-oxo-dG levels and also evaluated promoter methylation of 11 tumor suppressor genes (TSGs) in 128 liver biopsy samples. These biopsies were taken from patients with chronic hepatitis C, a condition causing oxidative damages in the liver. Among 5 factors evaluated, only increased levels of 8-oxo-dG was highly correlated with promoter methylation of TSGs (p<0.0001). This promoter methylation could have reduced expression of these tumor suppressor genes and contributed to carcinogenesis.
Mutagenesis
Yasui et al.[17] examined the fate of 8-oxo-dG when this oxidized derivative of deoxyguanosine was inserted into the thymidine kinase gene in a chromosome within human lymphoblastoid cells in culture. They inserted 8-oxo-dG into about 800 cells, and could detect the products that occurred after the insertion of this altered base, as determined from the clones produced after growth of the cells. 8-oxo-dG was restored to G in 86% of the clones, probably reflecting accurate base excision repair or translesion synthesis without mutation. G:C to T:A transversions occurred in 5.9% of the clones, single base deletions in 2.1% and G:C to C:G transversions in 1.2%. Together, these more common mutations totaled 9.2% of the 14% of mutations generated at the site of the 8-oxo-dG insertion. Among the other mutations in the 800 clones analyzed, there were also 3 larger deletions, of sizes 6, 33 and 135 base pairs. Thus 8-oxo-dG, if not repaired, can directly cause frequent mutations, some of which may contribute to carcinogenesis.
Role of DNA oxidation in gene regulation
As reviewed by Wang et al.,[18] oxidized guanine appears to have multiple regulatory roles in gene expression. As noted by Wang et al.,[18] genes prone to be actively transcribed are densely distributed in high GC-content regions of the genome. They then described three modes of gene regulation by DNA oxidation at guanine. In one mode, it appears that oxidative stress may produce 8-oxo-dG in a promoter of a gene. The oxidative stress may also inactivate OGG1. The inactive OGG1, which no longer excises 8-oxo-dG, nevertheless targets and complexes with 8-oxo-dG, and causes a sharp (~70o) bend in the DNA. This allows the assembly of a transcriptional initiation complex, up-regulating transcription of the associated gene. The experimental basis establishing this mode was also reviewed by Seifermann and Epe[19]
A second mode of gene regulation by DNA oxidation at a guanine,[18][20] occurs when an 8-oxo-dG is formed in a guanine rich, potential G-quadruplex-forming sequence (PQS) in the coding strand of a promoter, after which active OGG1 excises the 8-oxo-dG and generates an apurinic/apyrimidinic site (AP site). The AP site enables melting of the duplex to unmask the PQS, adopting a G-quadruplex fold (G4 structure/motif) that has a regulatory role in transcription activation.
A third mode of gene regulation by DNA oxidation at a guanine,[18] occurs when 8-oxo-dG is complexed with OGG1 and then recruits chromatin remodelers to modulate gene expression. Chromodomain helicase DNA-binding protein 4 (CHD4), a component of the (NuRD) complex, is recruited by OGG1 to oxidative DNA damage sites. CHD4 then attracts DNA and histone methylating enzymes that repress transcription of associated genes.
Seifermann and Epe[19] noted that the highly selective induction of 8-oxo-dG in the promoter sequences observed in transcription induction may be difficult to explain as a consequence of general oxidative stress. However, there appears to be a mechanism for site-directed generation of oxidized bases in promoter regions. Perillo et al.,[21][22] showed that the lysine-specific histone demethylase LSD1 generates a local burst of reactive oxygen species (ROS) that induces oxidation of nearby nucleotides when carrying out its function. As a specific example, after treatment of cells with an estrogen, LSD1 produced H2O2 as a by-product of its enzymatic activity. The oxidation of DNA by LSD1 in the course of the demethylation of histone H3 at lysine 9 was shown to be required for the recruitment of OGG1 and also topoisomerase IIβ to the promoter region of bcl-2, an estrogen-responsive gene, and subsequent transcription initiation.
8-oxo-dG does not occur randomly in the genome. In mouse embryonic fibroblasts, a 2 to 5-fold enrichment of 8-oxo-dG was found in genetic control regions, including promoters, 5'-untranslated regions and 3'-untranslated regions compared to 8-oxo-dG levels found in gene bodies and in intergenic regions.[23] In rat pulmonary artery endothelial cells, when 22,414 protein-coding genes were examined for locations of 8-oxo-dG, the majority of 8-oxo-dGs (when present) were found in promoter regions rather than within gene bodies.[24] Among hundreds of genes whose expression levels were affected by hypoxia, those with newly acquired promoter 8-oxo-dGs were upregulated, and those genes whose promoters lost 8-oxo-dGs were almost all downregulated.[24]
Positive role of 8-oxo-dG in memory
Oxidation of guanine, particularly within CpG sites, may be especially important in learning and memory. Methylation of cytosines occurs at 60–90% of CpG sites depending on the tissue type.[25] In the mammalian brain, ~62% of CpGs are methylated.[25] Methylation of CpG sites tends to stably silence genes.[26] More than 500 of these CpG sites are de-methylated in neuron DNA during memory formation and memory consolidation in the hippocampus[27][28] and cingulate cortex[28] regions of the brain. As indicated below, the first step in de-methylation of methylated cytosine at a CpG site is oxidation of the guanine to form 8-oxo-dG.
Role of oxidized guanine in DNA de-methylation
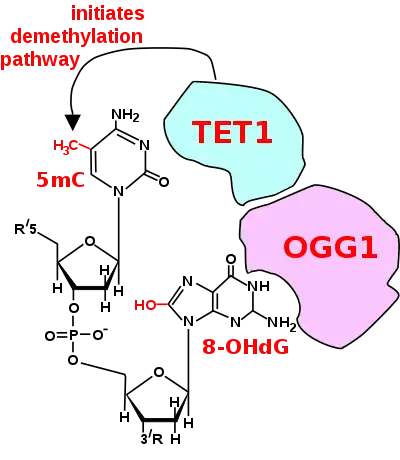
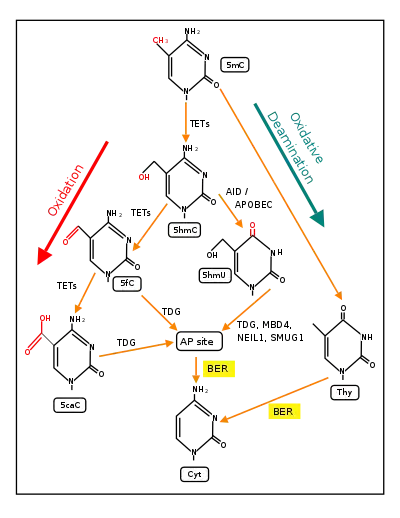
The first figure in this section shows a CpG site where the cytosine is methylated to form 5-methylcytosine (5mC) and the guanine is oxidized to form 8-oxo-2'-deoxyguanosine (in the figure this is shown in the tautomeric form 8-OHdG). When this structure is formed, the base excision repair enzyme OGG1 targets 8-OHdG and binds to the lesion without immediate excision. OGG1, present at a 5mCp-8-OHdG site recruits TET1, and TET1 oxidizes the 5mC adjacent to the 8-OHdG. This initiates de-methylation of 5mC.[29] TET1 is a key enzyme involved in de-methylating 5mCpG. However, TET1 is only able to act on 5mCpG if the guanine was first oxidized to form 8-hydroxy-2'-deoxyguanosine (8-OHdG or its tautomer 8-oxo-dG), resulting in a 5mCp-8-OHdG dinucleotide (see first figure in this section).[29] This initiates the de-methylation pathway on the methylated cytosine, finally resulting in an unmethylated cytosine, shown in the second figure in this section.
Altered protein expression in neurons, due to changes in methylation of DNA, (likely controlled by 8-oxo-dG-dependent de-methylation of CpG sites in gene promoters within neuron DNA) has been established as central to memory formation.[31]
Neurological conditions
Bipolar disorder
Evidence that oxidative stress induced DNA damage plays a role in bipolar disorder has been reviewed by Raza et al.[32] Bipolar patients have elevated levels of oxidatively induced DNA base damages even during periods of stable mental state.[33] The level of the base excision repair enzyme OGG1 that removes certain oxidized bases from DNA is also reduced compared to healthy individuals.[33]
Depressive disorder
Major depressive disorder is associated with an increase in oxidative DNA damage.[32] Increases in oxidative modifications of purines and pyrimidines in depressive patients may be due to impaired repair of oxidative DNA damages.[34]
Schizophrenia
Postmortem studies of elderly patients with chronic schizophrenia showed that oxidative DNA damage is increased in the hippocampus region of the brain.[35] The mean proportion of neurons with the oxidized DNA base 8-oxo-dG was 10-fold higher in patients with schizophrenia than in comparison individuals. Evidence indicating a role of oxidative DNA damage in schizophrenia has been reviewed by Raza et al.[32] and Markkanen et al.[36]
RNA Oxidation
RNAs in native milieu are exposed to various insults. Among these threats, oxidative stress is one of the major causes of damage to RNAs. The level of oxidative stress that a cell endures is reflected by the quantity of reactive oxygen species (ROS). ROS are generated from normal oxygen metabolism in cells and are recognized as a list of active molecules, such as O2•−, 1O2, H2O2 and, •OH .[37] A nucleic acid can be oxidized by ROS through a Fenton reaction.[38] To date, around 20 oxidative lesions have been discovered in DNA.[39] RNAs are likely to be more sensitive to ROS for the following reasons: i) the basically single-stranded structure exposes more sites to ROS; ii) compared with nuclear DNA, RNAs are less compartmentalized; iii) RNAs distribute broadly in cells not only in the nucleus as DNAs do, but also in large portions in the cytoplasm.[40][41] This theory has been supported by a series of discoveries from rat livers, human leukocytes, etc. Actually, monitoring a system by applying the isotopical label [18O]-H2O2 shows greater oxidation in cellular RNA than in DNA. Oxidation randomly damages RNAs, and each attack bring problems to the normal cellular metabolism. Although alteration of genetic information on mRNA is relatively rare, oxidation on mRNAs in vitro and in vivo results in low translation efficiency and aberrant protein products.[42] Though the oxidation strikes the nucleic strands randomly, particular residues are more susceptible to ROS, such hotspot sites being hit by ROS at a high rate. Among all the lesions discovered thus far, one of the most abundant in DNA and RNA is the 8-hydroxyguanine.[43] Moreover, 8-hydroxyguanine is the only one measurable among all the RNA lesions. Besides its abundance, 8-hydroxydeoxyguanosine (8-oxodG) and 8-hydroxyguanosine (8-oxoG) are identified as the most detrimental oxidation lesions for their mutagenic effect,[44] in which this non-canonical counterpart can faultily pair with both adenine and cytosine at the same efficiency.[45][46] This mis-pairing brings about the alteration of genetic information through the synthesis of DNA and RNA. In RNA, oxidation levels are mainly estimated through 8-oxoG-based assays. So far, approaches developed to directly measure 8-oxoG level include HPLC-based analysis and assays employing monoclonal anti-8-oxoG antibody. The HPLC-based method measures 8-oxoG with an electrochemical detector (ECD) and total G with a UV detector.[47] The ratio that results from comparing the two numbers provides the extent that the total G is oxidized. Monoclonal anti-8-oxoG mouse antibody is broadly applied to directly detect this residue on either tissue sections or membrane, offering a more visual way to study its distribution in tissues and in discrete subsets of DNA or RNA. The established indirect techniques are mainly grounded on this lesion’s mutagenic aftermath, such as the lacZ assay.[48] This method was first set up and described by Taddei and was a potentially powerful tool to understand the oxidation situation at both the RNA sequence level and single nucleotide level. Another source of oxidized RNAs is mis-incorporation of oxidized counterpart of single nucleotides. Indeed, the RNA precursor pool size is hundreds of sizes bigger than DNA’s.
Potential factors for RNA quality control
There have been furious debates on whether the issue of RNA quality control does exist. However, with the concern of various lengths of half lives of diverse RNA species ranging from several minutes to hours, degradation of defective RNA can not easily be attributed to its transient character anymore. Indeed, reaction with ROS takes only few minutes, which is even shorter than the average life-span of the most unstable RNAs.[40] Adding the fact that stable RNA take the lion’s share of total RNA, RNA error deleting becomes hypercritical and should not be neglected anymore. This theory is upheld by the fact that level of oxidized RNA decreases after removal of the oxidative challenge.[49][50] Some potential factors include ribonucleases, which are suspected to selectively degrade damaged RNAs under stresses. Also enzymes working at RNA precursor pool level, are known to control quality of RNA sequence by changing error precursor to the form that can't be included directly into nascent strand.
References
- Burrows CJ, Muller JG (May 1998). "Oxidative Nucleobase Modifications Leading to Strand Scission". Chem. Rev. 98 (3): 1109–1152. doi:10.1021/cr960421s. PMID 11848927.
- Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB (December 2010). "Oxidative stress, inflammation, and cancer: how are they linked?". Free Radic. Biol. Med. 49 (11): 1603–16. doi:10.1016/j.freeradbiomed.2010.09.006. PMC 2990475. PMID 20840865.
- Massaad CA, Klann E (May 2011). "Reactive oxygen species in the regulation of synaptic plasticity and memory". Antioxid. Redox Signal. 14 (10): 2013–54. doi:10.1089/ars.2010.3208. PMC 3078504. PMID 20649473.
- Beckhauser TF, Francis-Oliveira J, De Pasquale R (2016). "Reactive Oxygen Species: Physiological and Physiopathological Effects on Synaptic Plasticity". J Exp Neurosci. 10 (Suppl 1): 23–48. doi:10.4137/JEN.S39887. PMC 5012454. PMID 27625575.
- Cooke MS, Evans MD, Dizdaroglu M, Lunec J (2003). "Oxidative DNA damage: mechanisms, mutation, and disease". FASEB J. 17 (10): 1195–214. CiteSeerX 10.1.1.335.5793. doi:10.1096/fj.02-0752rev. PMID 12832285. S2CID 1132537.
- Dizdaroglu M (1992). "Oxidative damage to DNA in mammalian chromatin". Mutat. Res. 275 (3–6): 331–42. doi:10.1016/0921-8734(92)90036-o. PMID 1383774.
- Hamilton ML, Guo Z, Fuller CD, Van Remmen H, Ward WF, Austad SN, Troyer DA, Thompson I, Richardson A (2001). "A reliable assessment of 8-oxo-2-deoxyguanosine levels in nuclear and mitochondrial DNA using the sodium iodide method to isolate DNA". Nucleic Acids Res. 29 (10): 2117–26. doi:10.1093/nar/29.10.2117. PMC 55450. PMID 11353081.
- Swenberg JA, Lu K, Moeller BC, Gao L, Upton PB, Nakamura J, Starr TB (2011). "Endogenous versus exogenous DNA adducts: their role in carcinogenesis, epidemiology, and risk assessment". Toxicol Sci. 120 (Suppl 1): S130–45. doi:10.1093/toxsci/kfq371. PMC 3043087. PMID 21163908.
- Prasad AR, Prasad S, Nguyen H, Facista A, Lewis C, Zaitlin B, Bernstein H, Bernstein C (2014). "Novel diet-related mouse model of colon cancer parallels human colon cancer". World J Gastrointest Oncol. 6 (7): 225–43. doi:10.4251/wjgo.v6.i7.225. PMC 4092339. PMID 25024814.
- Valavanidis A, Vlachogianni T, Fiotakis K, Loridas S (2013). "Pulmonary oxidative stress, inflammation and cancer: respirable particulate matter, fibrous dusts and ozone as major causes of lung carcinogenesis through reactive oxygen species mechanisms". Int J Environ Res Public Health. 10 (9): 3886–907. doi:10.3390/ijerph10093886. PMC 3799517. PMID 23985773.
- Tsuei J, Chau T, Mills D, Wan YJ (Nov 2014). "Bile acid dysregulation, gut dysbiosis, and gastrointestinal cancer". Exp Biol Med (Maywood). 239 (11): 1489–504. doi:10.1177/1535370214538743. PMC 4357421. PMID 24951470.
- Ajouz H, Mukherji D, Shamseddine A (2014). "Secondary bile acids: an underrecognized cause of colon cancer". World J Surg Oncol. 12: 164. doi:10.1186/1477-7819-12-164. PMC 4041630. PMID 24884764.
- Bernstein C, Bernstein H (2015). "Epigenetic reduction of DNA repair in progression to gastrointestinal cancer". World J Gastrointest Oncol. 7 (5): 30–46. doi:10.4251/wjgo.v7.i5.30. PMC 4434036. PMID 25987950.
- Scott TL, Rangaswamy S, Wicker CA, Izumi T (2014). "Repair of oxidative DNA damage and cancer: recent progress in DNA base excision repair". Antioxid. Redox Signal. 20 (4): 708–26. doi:10.1089/ars.2013.5529. PMC 3960848. PMID 23901781.
- Li J, Braganza A, Sobol RW (2013). "Base excision repair facilitates a functional relationship between Guanine oxidation and histone demethylation". Antioxid. Redox Signal. 18 (18): 2429–43. doi:10.1089/ars.2012.5107. PMC 3671628. PMID 23311711.
- Nishida N, Arizumi T, Takita M, Kitai S, Yada N, Hagiwara S, Inoue T, Minami Y, Ueshima K, Sakurai T, Kudo M (2013). "Reactive oxygen species induce epigenetic instability through the formation of 8-hydroxydeoxyguanosine in human hepatocarcinogenesis". Dig Dis. 31 (5–6): 459–66. doi:10.1159/000355245. PMID 24281021.
- Yasui M, Kanemaru Y, Kamoshita N, Suzuki T, Arakawa T, Honma M (2014). "Tracing the fates of site-specifically introduced DNA adducts in the human genome". DNA Repair (Amst.). 15: 11–20. doi:10.1016/j.dnarep.2014.01.003. PMID 24559511.
- Wang R, Hao W, Pan L, Boldogh I, Ba X (October 2018). "The roles of base excision repair enzyme OGG1 in gene expression". Cell. Mol. Life Sci. 75 (20): 3741–3750. doi:10.1007/s00018-018-2887-8. PMC 6154017. PMID 30043138.
- Seifermann M, Epe B (June 2017). "Oxidatively generated base modifications in DNA: Not only carcinogenic risk factor but also regulatory mark?". Free Radic. Biol. Med. 107: 258–265. doi:10.1016/j.freeradbiomed.2016.11.018. PMID 27871818.
- Fleming AM, Burrows CJ (August 2017). "8-Oxo-7,8-dihydroguanine, friend and foe: Epigenetic-like regulator versus initiator of mutagenesis". DNA Repair (Amst.). 56: 75–83. doi:10.1016/j.dnarep.2017.06.009. PMC 5548303. PMID 28629775.
- Perillo B, Di Santi A, Cernera G, Ombra MN, Castoria G, Migliaccio A (2014). "Nuclear receptor-induced transcription is driven by spatially and timely restricted waves of ROS. The role of Akt, IKKα, and DNA damage repair enzymes". Nucleus. 5 (5): 482–91. doi:10.4161/nucl.36274. PMC 4164490. PMID 25482200.
- Perillo B, Ombra MN, Bertoni A, Cuozzo C, Sacchetti S, Sasso A, Chiariotti L, Malorni A, Abbondanza C, Avvedimento EV (January 2008). "DNA oxidation as triggered by H3K9me2 demethylation drives estrogen-induced gene expression". Science. 319 (5860): 202–6. Bibcode:2008Sci...319..202P. doi:10.1126/science.1147674. PMID 18187655. S2CID 52330096.
- Ding Y, Fleming AM, Burrows CJ (February 2017). "Sequencing the Mouse Genome for the Oxidatively Modified Base 8-Oxo-7,8-dihydroguanine by OG-Seq". J. Am. Chem. Soc. 139 (7): 2569–2572. doi:10.1021/jacs.6b12604. PMC 5440228. PMID 28150947.
- Pastukh V, Roberts JT, Clark DW, Bardwell GC, Patel M, Al-Mehdi AB, Borchert GM, Gillespie MN (December 2015). "An oxidative DNA "damage" and repair mechanism localized in the VEGF promoter is important for hypoxia-induced VEGF mRNA expression". Am. J. Physiol. Lung Cell Mol. Physiol. 309 (11): L1367–75. doi:10.1152/ajplung.00236.2015. PMC 4669343. PMID 26432868.
- Fasolino M, Zhou Z (May 2017). "The Crucial Role of DNA Methylation and MeCP2 in Neuronal Function". Genes (Basel). 8 (5): 141. doi:10.3390/genes8050141. PMC 5448015. PMID 28505093.
- Bird A (January 2002). "DNA methylation patterns and epigenetic memory". Genes Dev. 16 (1): 6–21. doi:10.1101/gad.947102. PMID 11782440.
- Duke CG, Kennedy AJ, Gavin CF, Day JJ, Sweatt JD (July 2017). "Experience-dependent epigenomic reorganization in the hippocampus". Learn. Mem. 24 (7): 278–288. doi:10.1101/lm.045112.117. PMC 5473107. PMID 28620075.
- Halder R, Hennion M, Vidal RO, Shomroni O, Rahman RU, Rajput A, Centeno TP, van Bebber F, Capece V, Garcia Vizcaino JC, Schuetz AL, Burkhardt S, Benito E, Navarro Sala M, Javan SB, Haass C, Schmid B, Fischer A, Bonn S (January 2016). "DNA methylation changes in plasticity genes accompany the formation and maintenance of memory". Nat. Neurosci. 19 (1): 102–10. doi:10.1038/nn.4194. PMC 4700510. PMID 26656643.
- Zhou X, Zhuang Z, Wang W, He L, Wu H, Cao Y, Pan F, Zhao J, Hu Z, Sekhar C, Guo Z (September 2016). "OGG1 is essential in oxidative stress induced DNA demethylation". Cell. Signal. 28 (9): 1163–71. doi:10.1016/j.cellsig.2016.05.021. PMID 27251462.
- Bayraktar G, Kreutz MR (2018). "The Role of Activity-Dependent DNA Demethylation in the Adult Brain and in Neurological Disorders". Front Mol Neurosci. 11: 169. doi:10.3389/fnmol.2018.00169. PMC 5975432. PMID 29875631.
- Day JJ, Sweatt JD (November 2010). "DNA methylation and memory formation". Nat. Neurosci. 13 (11): 1319–23. doi:10.1038/nn.2666. PMC 3130618. PMID 20975755.
- Raza MU, Tufan T, Wang Y, Hill C, Zhu MY (August 2016). "DNA Damage in Major Psychiatric Diseases". Neurotox Res. 30 (2): 251–67. doi:10.1007/s12640-016-9621-9. PMC 4947450. PMID 27126805.
- Ceylan D, Tuna G, Kirkali G, Tunca Z, Can G, Arat HE, Kant M, Dizdaroglu M, Özerdem A (May 2018). "Oxidatively-induced DNA damage and base excision repair in euthymic patients with bipolar disorder". DNA Repair (Amst.). 65: 64–72. doi:10.1016/j.dnarep.2018.03.006. PMC 7243967. PMID 29626765.
- Czarny P, Kwiatkowski D, Kacperska D, Kawczyńska D, Talarowska M, Orzechowska A, Bielecka-Kowalska A, Szemraj J, Gałecki P, Śliwiński T (February 2015). "Elevated level of DNA damage and impaired repair of oxidative DNA damage in patients with recurrent depressive disorder". Med. Sci. Monit. 21: 412–8. doi:10.12659/MSM.892317. PMC 4329942. PMID 25656523.
- Nishioka N, Arnold SE (2004). "Evidence for oxidative DNA damage in the hippocampus of elderly patients with chronic schizophrenia". Am J Geriatr Psychiatry. 12 (2): 167–75. doi:10.1097/00019442-200403000-00008. PMID 15010346.
- Markkanen E, Meyer U, Dianov GL (June 2016). "DNA Damage and Repair in Schizophrenia and Autism: Implications for Cancer Comorbidity and Beyond". Int J Mol Sci. 17 (6): 856. doi:10.3390/ijms17060856. PMC 4926390. PMID 27258260.
- Buechter, DD. (1988) Free radicals and oxygen toxicity.Pharm Res. 5:253-60.
- Wardman, P. and Candeias, L.P. (1996). Fenton chemistry: an introduction. Radiat. Res. 145, 523–531.
- Cooke MS, Evans MD, Dizdaroglu M, Lunec J (2003). "Oxidative DNA damage: mechanisms, mutation, and disease". FASEB J. 17 (10): 195–1214. doi:10.1096/fj.02-0752rev. PMID 12832285. S2CID 1132537.
- Li Z, Wu J, Deleo CJ (2006). "RNA Damage and Surveillance under Oxidative Stress". IUBMB Life. 58 (10): 581–588. doi:10.1080/15216540600946456. PMID 17050375. S2CID 30141613.
- Hofer T, Seo AY, Prudencio M, Leeuwenburgh C (2006). "A method to determine RNA and DNA oxidation simultaneously by HPLC-ECD: greater RNA than DNA oxidation in rat liver after doxorubicin administration". Biol. Chem. 387: 103–111. doi:10.1515/bc.2006.014. PMID 16497170. S2CID 13613547.
- Dukan S, Farwell A, Ballesteros M, Taddei F, Radman M, Nystrom T (2000). "Protein oxidation in response to increased transcriptional and translational errors". Proc. Natl. Acad. Sci. USA. 97 (11): 5746–5749. Bibcode:2000PNAS...97.5746D. doi:10.1073/pnas.100422497. PMC 18504. PMID 10811907.
- Gajewski E, Rao G, Nackerdien Z, Dizdaroglu M (1990). "Modification of DNA bases in mammalian chromatin by radiationgenerated free radicals". Biochemistry. 29 (34): 7876–7882. doi:10.1021/bi00486a014. PMID 2261442.
- Ames BN, Gold LS (1991). "Endogenous mutagens and the causes of aging and cancer". Mutat. Res. 250 (1–2): 3–16. doi:10.1016/0027-5107(91)90157-j. PMID 1944345.
- Shibutani S, Takeshita M, Grollman AP (1991). "Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG". Nature. 349 (6308): 431–434. Bibcode:1991Natur.349..431S. doi:10.1038/349431a0. PMID 1992344. S2CID 4268788.
- Taddei F, Hayakawa H, Bouton M, Cirinesi A, Matic I, Sekiguchi M, Radman M (1997). "Counteraction by MutT protein of transcriptional errors caused by oxidative damage". Science. 278 (5335): 128–130. doi:10.1126/science.278.5335.128. PMID 9311918.
- Weimann A, Belling D, Poulsen HE (2002). "Quantification of 8-oxoGuanine and guanine as the nucleobase, nucleoside and deoxynucleoside forms in human urine by high-performance liquid chromatography-electrospray tandem mass spectrometry". Nucleic Acids Res. 30 (2): E7. doi:10.1093/nar/30.2.e7. PMC 99846. PMID 11788733.
- Park EM, Shigenaga MK, Degan P, Korn TS, Kitzler JW, Wehr CM, Kolachana P, Ames BN (1992). "Assay of excised oxidative DNA lesions: isolation of 8-oxoguanine and its nucleoside derivatives from biological fluids with a monoclonal antibody column". Proc. Natl. Acad. Sci. USA. 89 (8): 3375–3379. Bibcode:1992PNAS...89.3375P. doi:10.1073/pnas.89.8.3375. PMID 1565629.
- Shen Z, Wu W, Hazen SL (2000). "Activated leukocytes oxidatively damage DNA, RNA, and the nucleotide pool through halide-dependent formation of hydroxyl radical". Biochemistry. 39: 5474–5482. doi:10.1021/bi992809y. PMID 10820020.
- Kajitani K, Yamaguchi H, Dan Y, Furuichi M, Kang D, Nakabeppu Y (2006). "MTH1, and oxidized purine nucleoside triphosphatase, suppresses the accumulation of oxidative damage of nucleic acids in the hippocampal microglia during kainite-induced excitotoxicity". J. Neurosci. 26 (6): 1688–1689. doi:10.1523/jneurosci.4948-05.2006. PMC 6793619. PMID 16467516.