Dicarboxylic aminoaciduria
Dicarboxylic aminoaciduria is a rare form of aminoaciduria (1:35 000 births[2]) which is an autosomal recessive disorder of urinary glutamate and aspartate due to genetic errors related to transport of these amino acids.[3] Mutations resulting in a lack of expression of the SLC1A1 gene, a member of the solute carrier family, are found to cause development of dicarboxylic aminoaciduria in humans. SLC1A1 encodes for EAAT3 which is found in the neurons, intestine, kidney, lung, and heart.[3][4] EAAT3 is part of a family of high affinity glutamate transporters which transport both glutamate and aspartate across the plasma membrane.
| Dicarboxylic aminoaciduria | |
|---|---|
| Other names | Glutamate-aspartate transport defect[1] |
| Specialty | Endocrinology |
Symptoms
Dicarboxylic aminoaciduria involves excretion of urinary glutamate and aspartate, resulting from the incomplete reabsorption of anionic amino acids from the glomerular filtrate in the kidney.[3] This affects a diseased individual's amino acid pool, as they will have to spend additional resources to replenish the amino acids which would have otherwise been present. Additionally, glutamate transporters are responsible for the synaptic release of the glutamate (neurotransmitter) within the interneuronal synaptic cleft. This hindrance of functionality in individuals with dicarboxylic aminoaciduria may be related to growth retardation, intellectual disability, and a tendency toward fasting hypoglycemia and ketoacidosis.[3][5] Dicarboxylic aminoaciduria is diagnosed by finding the increased presence of glutamate and aspartate in the urine.[3]
Cause
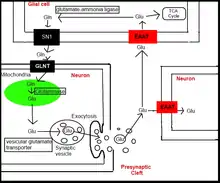
Glutamate transporters are proficient at pumping glutamate into cells due to their ability to couple with inorganic ions.[4] Transport of glutamate into the cell requires the coupling of three sodium ions as well as a proton, whereas transport out of the cell requires a single potassium ion.[4] This transport results in two positive charges being displaced across the membrane per cycle.[4] Moreover, this process is dependent on a pH gradient, due to glutamate needing to be protenated prior to transport.[4]
Mutations
Dicarboxylic aminoaciduria is the result of a point mutation of tryptophan to arginine at position 445 and a deletion mutation of isoleucine at position 395.[3] EAAT3 is found in location 9p24, it is primarily expressed in the brain and kidneys.[6]
Metabolism
In the gastrointestinal tract, protein digestion and absorption are key to establishing and maintaining amino acid pools. In the case of dicarboxylic aminoaciduria, where glutamate and aspartate transport are impaired, the alanine, aspartate and glutamate metabolism are affected. Enterocytes in the intestines break up peptides into residual amino acids where they would normally use charge-specific amino acid transporters to get across the epithelial cells. In dicarboxylic aminoaciduria, the anionic amino acid transporter, EAAT3, cannot bring glutamate and aspartate across epithelial cells, leading to them being excreted via the urine.
Examples
Below is an example of how glutamate is used to synthesize alanine via alanine transaminase.
Another example is the conversion of aspartate to glutamate via the enzyme aspartate transaminase.
- Aspartate + α-ketoglutarate ⇌ Oxaloacetate + Glutamate
.
References
- "Dicarboxylic aminoaciduria | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Retrieved 16 April 2019.
- Camargo SM, Bockenhauer D, Kleta R (April 2008). "Aminoacidurias: Clinical and molecular aspects". Kidney Int. 73 (8): 918–25. doi:10.1038/sj.ki.5002790. PMID 18200002.
- Bailey CG, Ryan RM, Thoeng AD, Ng C, King K, Vanslambrouck JM, Auray-Blais C, Vandenberg RJ, Bröer S, Rasko JE (January 2011). "Loss-of-function mutations in the glutamate transporter SLC1A1 cause human dicarboxylic aminoaciduria". J. Clin. Invest. 121 (1): 446–53. doi:10.1172/JCI44474. PMC 3007158. PMID 21123949.
- Hediger MA (October 1999). "Glutamate transporters in kidney and brain". Am. J. Physiol. 277 (4 Pt 2): F487–92. doi:10.1152/ajprenal.1999.277.4.F487. PMID 10516270.
- Bröer S. (January 2008). "Amino acid transport across mammalian intestinal and renal epithelia". Physiol. Rev. 88 (1): 249–86. doi:10.1152/physrev.00018.2006. PMID 18195088.
- Smith CP, Weremowicz S, Kanai Y, Stelzner M, Morton CC, Hediger MA (March 1994). "Assignment of the gene coding for the human high-affinity glutamate transporter EAAC1 to 9p24: potential role in dicarboxylic aminoaciduria and neurodegenerative disorders". Genomics. 20 (2): 335–336. doi:10.1006/geno.1994.1183. PMID 8020993.
External links
| Classification |
|---|