Discovery and development of beta2 agonists
β2-adrenoceptor agonists is a group of drugs that act selectively on β2-receptors in the lungs causing bronchodilation. β2-agonists are used to treat asthma and COPD, diseases that cause obstruction in the airways. Prior to their discovery, the non-selective beta-agonist isoprenaline was used. The aim of the drug development through the years has been to minimise side effects, achieve selectivity and longer duration of action. The mechanism of action is well understood and has facilitated the development. The structure of the binding site and the nature of the binding is also well known, as is the structure activity relationship.
History
The β2-selective agonists were developed in the 20th century and are a very valuable class of drugs. In 1901 Jōkichi Takamine isolated the hormone adrenalin, also known as epinephrine.[1] In 1890 adrenalin was first given to asthma patients orally. It had little or no effect because it is metabolized in the digestive tract and is deactivated. In 1930 epinephrine was for the first time given subcutaneously and was discovered to have a positive effect on asthma.[1] When given subcutaneously adrenalin affects the whole body, giving various side effects and thus reducing the value of this treatment. The inhaled route was later tried and it gave much less adverse effects, but still had inconvenient side effects like fear, anxiety, restlessness, headache, dizziness and palpitation.[1]
In 1940 isoproterenol (isoprenaline) was discovered. This compound had a similar effect as adrenalin but fewer side effects were found. In 1949 isoproterenol was used generally to treat asthma patients, given sublingually or inhaled.[1] The first pressurized metered-dose inhaler was introduced in 1956. This was much more convenient for patients than the previously used squeeze-bulb inhalers. The pressurized metered-dose inhaler technique developed rapidly in the 1970s.
In 1967, it was shown that the β2-receptor was responsible for bronchodilation and this led to development of more selective drugs.[1] In 1961 orciprenaline, a longer acting β2-agonist was found, but it was not as potent as isoproterenol. Orciprenaline does not have the catechol structure which was the reason for the longer action time. In the mid-1960s, albuterol or salbutamol was discovered, followed by tributalin and fenoterol a few years later. Albuterol and terbutaline gave fewer side effects, such as increased heart rate, than isoproterenol. The pharmaceutical company Glaxo discovered salmeterol, a long-acting β2-agonist that had bronchodilation activity for up to 12 hours. It was marketed in 1990. Formoterol, another long-acting β2-agonist, was marketed shortly after. This long duration of action made the treatment for severe asthma and COPD more convenient for the patients because it is inhaled twice a day.[1] In 2013 an extra long-acting β2-agonist, vilanterol, was marketed. Its duration of action lasts for 24 hours which should improve patients' compliance and make the treatment more convenient.[2]
Clinical use
Asthma
β2-agonists are used to treat asthma, an inflammatory disease in the airways. The inflammation makes the bronchia sensitive to some factors in the environment, which causes bronchoconstriction. When this constriction occurs, the airways get narrow and it causes symptoms like wheezing, chest tightness, shortness of breath, and coughing. The muscles in the airways tighten, and cells in the airway start to produce more mucus than usual, which narrows the airways even more. The symptoms often start in childhood, but it can start at any age .[3]
Chronic obstructive pulmonary disease
Both short- and long-acting β2-agonists are used to treat chronic obstructive pulmonary disease. COPD causes airflow limitations in the lungs because of inflammation. Smoking is the main risk factor but inhalation of toxic and harmful particles and gases can also cause the disease. The symptoms are abnormal mucus production, inflation in the lungs that causes airflow limitation, abnormal gas exchange and pulmonary hypertension. COPD is most common in people over fifty who have a long history of smoking. The symptoms are at first mild but worsen over time.[4]
Use
There are two types of β2-agonists, long- and short-acting. They are both inhaled and given by aerosol delivery devices.[5][6] Long-lasting β2-agonists are often given in a combination with corticosteroids to treat asthma. Short-acting β2-agonists are used to treat exercise-induced asthma, [7] and for asthma patients to get a quick relief of symptoms. They are taken 10–15 minutes before exercise. The bronchodilation begins few minutes after inhalation of short-acting β2-agonists and lasts from 4 to 8 hours.[8] [9] Long-lasting β2-agonists are discouraged in treating acute exercise-induced asthma, because their chronic use might mask poorly controlled asthma.[10]
Mechanism of Action
Pharmacokinetic
The kinetics of airway smooth muscle relaxation, as long as the onset and duration of bronchodilation in asthmatic patients, are reflected by the difference in the mechanism of interaction of short- (SABAs) and longacting β2-agonists (LABAs) and the β2-receptor.[11] There are many formulations of selective β2-agonists; inhalation is the route of choice because it is the most rapidly effective and is associated with minimum side effects.[12] Sulfate conjugates are the main metabolites; protein binding is rather weak and only insignificant interactions have been found with other drugs.[13] The main enzymes that regulate metabolism of the catecholamines are COMT and MAO. The commercial SABAs, salbutamol and terbutaline are resistant to COMT but are slowly metabolised by MAO, while the LABAs are resistant to both COMT and MAO. Also, the long duration of action for salmeterol is related to increased lipophilicity of the molecules, allowing it to remain for a longer time in the lungs.[14] β2-agonists are mainly eliminated by the renal process after parenteral administration, while after oral administration a more pronounced metabolic clearance (high first pass effect) is responsible for a low bioavailability. The elimination after inhalation has not been studied but the profile is likely somewhere between what we see after parenteral and oral administration.[13]
Binding to β adrenergic receptors
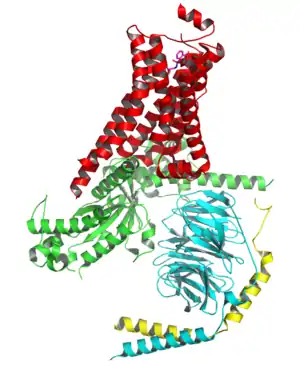
β-receptors are membrane-bound receptors coupled to G-proteins. Three types of β-receptors have been identified by molecular pharmacology. β1 receptors make up to 75% of all beta receptors and are predominantly located in the heart. β2 receptors are found in vascular and bronchial smooth muscle. β3 receptors, which are presumed to be involved in fatty acid metabolism, are located in the adipocytes.[15]
G-protein coupled receptors consist of single polypeptide chains of 300-600 amino acids and span the plasma membrane seven times.[16] There are three extracellular loops, one of them being the amino-terminus, and three intracellular loops with a carboxy-terminus.[11] The hydrophilic pocket is formed within the membrane by the seven alpha-helical transmembrane domains. The ligand binds to the hydrophilic pocket in the receptor protein and activates the receptor, giving rise to the cellular effect.[16]
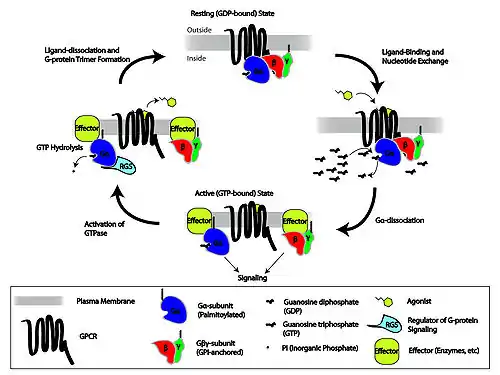
When the β2-agonist binds and activates the β2-adrenoreceptor intracellular signaling becomes largely affected through cyclic adenosine monophosphate (cAMP) and protein kinase A (PKA). The coupling of the β2-receptor to adenylate cyclase is affected through a trimeric Gs protein, as shown in figure 2, consisting of an α-subunit (which stimulates adenylate cyclase) and βγ-subunits (which transduce other signals). Adenylate cyclase catalyzes the conversion of adenosine triphosphate into cAMP, which is a second messenger, thereby increasing the intracellular cAMP levels, resulting in relaxation of smooth muscles. cAMP levels are regulated through the activity of phosphodiesterase isozymes/isoforms, which degrades it to 5′-AMP.[11][16] The mechanism by which cAMP induces relaxation in airway smooth muscle cells is not fully understood. It is believed that cAMP catalyzes the activation of PKA, which in turn phosphorylates key regulatory proteins involved in the control of muscle tone. cAMP also has a role in inhibition of calcium ion (Ca2+) release from intracellular stores, sequestration of intracellular Ca2+, and reduction of membrane Ca2+ entry, leading to relaxation of the airway smooth muscle.[11]
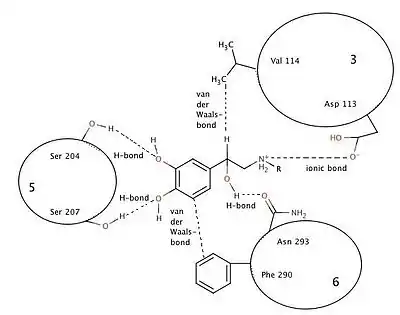
The binding of norephinephrine to the binding site consists of one ionic bond, three hydrogen bonds and van der Waals interaction in two places. Ionic bonding forms between the protonated amine and Asp-113 in helix 3. Hydrogen bonds are formed from the hydroxyl groups, linked to the catechol ring, to Ser-204 and Ser-207 in helix 5. This binding limits configurational and rotational freedom. Van der Waals forces between aromatic catechol ring and Phe-290 in helix 6 residue and Val-114 in helix 3 residue reinforce the binding. It is believed that the N-alkyl substituents fit into a hydrophobic pocket formed between residues in helix 6 and 7. The beta-carbon is chiral and must have the R-configuration so that the beta-hydroxyl group is oriented towards the Asn-293 residue in helix 6 to form a hydrogen bond essential to binding to the beta-2 receptor.[14]
Mechanism of long acting β2-agonists
Two theories explain the long time of action for LABAs. The first explained the long action in terms of a putative “exosite” or “exoceptor” distinct from the β2-adrenoreceptor that long aliphatic tail of salmeterol binds to with high affinity. This allows the active saligenin head to angle on and off the receptor to activate it repeatedly.[17] Formoterol does not have a long side chain like salmeterol to bind to the “exosite”, so this theory has been questioned. In 1994 Anderson. et al. introduced the plasmalemma diffusion microkinetic theory, explaining what happens to the β2- agonist in the cell membrane lipid bilayer and in the aqueous biophase closest to the binding site of the β2-adrenoceptor. It is postulated that the plasmalemma lipid bilayer of airway smooth muscles acts as a depot for β2-adrenoceptor agonists. β2-adrenoceptor agonists remain available to interact with the β2-adrenoceptor active site after having partitioned into the lipid bilayer.[17]
Structure Activity Relationships (SAR)
Basic structure of agonists
The fundamental pharmacophore for all adrenergic agonists is a substituted phenethylamine which increases the duration of action.[14]
Activity of β2-adrenoceptor agonists
Adrenergic agonists that are selective for the β2 subtype cause bronchial dilation and might be expected to relieve the bronchospasm of an asthmatic attack. Nonselective β-agonists have stimulatory cardiac effects and therefore have limited use in cardiac patients with asthma.[14]
Administration of higher doses of short-acting β2-agonists increases duration of action but also increases side effects such as cardiac effects. One approach to avoid these side effects is to use structurally different features that may minimize absorption into systemic circulation. For example, one could use drugs that transform into inactive metabolites upon entry into systemic circulation.[18]
Substituents on the pharmacophore influence whether an analog will be direct- or indirect-acting or a mixture of both. It also influences the specificity for the β-receptor subtypes. Direct-acting analog binds the β-adrenergic receptors directly and generates sympathetic response. Indirect-acting analog causes agonistic effect but without a direct binding to the β-adrenergic receptor, for example, by promoting release of norepinephrine (NE) from the presynaptic terminal or by inhibiting reuptake of released NE.[14]
Basic structure for every β2-agonists
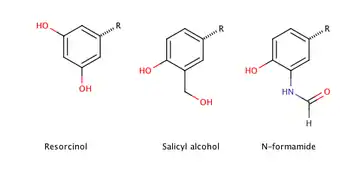
Figure 4 shows the phenyl rings that are used for β2-agonists. They are named resorcinol ring, a salicyl alcohol or an m-formamide group.[14] Figure 5 shows where different substituents on phenylethylamine take places marked as different R-groups.
Activity of R-groups listed
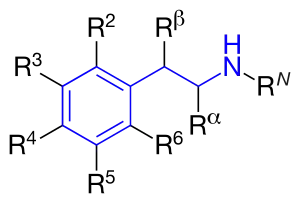
The difference between substituents R1-R5 is described below. All marketed β2-agonists have hydroxyl group in position R3 and most often in position R5.[14]
RN: This group determines α- or β-receptor selectivity. The larger the substituent, the greater the selectivity for the β-receptor. If t-butyl is positioned at RN it shows high affinity for the β2-receptor. A long phenylbutoxyhexyl substituent in this position gives high β2-selectivity and also high lipophilicity and therefore a longer duration of action.[14]
Rα: Substituents other than hydrogen would give increased duration of action. An ethyl group would increase the selectivity for the β2-receptor. However, an ethyl group seems to cause increased adverse effects and low β2-receptor potency compared to other β2-selective agonists.[14]
Rβ: A hydroxyl group gives direct action to the β-receptor. As noted earlier, all marketed β2-agonists have a hydroxyl group in this position which makes the compound chiral, and is active when it has the (R)-configuration.[14]
R5 or R3: Hydroxyl group placed on carbon number 5 or 3 (meta position) gives direct action to the β-adrenergic receptor.[14]
R4: Either hydroxyl group or hydrogen group in this position gives direct action to the β-receptor.[14]
Summarizing a few β2-adrenoceptor agonists and their structure activity shows how they act differently referred to potency, selectivity, affinity and duration of action (see table 1):
| Chemical structure | Name | Description |
|---|---|---|
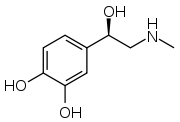 | Epinephrine (adrenalin) | Combination of catechol nucleus, β-hydroxyl group and N-methyl group. These idendities give direct action and a strong affinity for all adrenergic receptors.[14] |
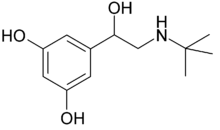 | Terbutaline | Combination of N-t-butyl and a resorcinol phenyl ring that gives potency to the β2-receptor.[14] |
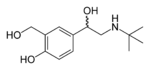 | Albuterol (Salbutamol) | Has N-t-butyl and a salicyl alcohol phenyl ring which gives it optimal β2-selectivity.[14] Salbutamol is a short-acting β2-agonist, but has a rapid onset of action.[18] Its onset by inhalation is within 5 minutes. The hydroxyl group on the β-carbon is a mixture of S- and R-isomers, where the R-isomer is the active one. Another drug, Levalbuterol, has exactly the same structure. it contains only the R-isomer of Albutarol and is therefore much more active and the dose would be fourfold less for Levalbuterol than Albutarol.[14] |
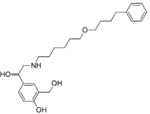 | Salmeterol | Has a N-phenylbutoxyhexyl substituent, β-hydroxyl group and a salicyl phenyl ring that gives potency and direct selectivity for β2-receptor.[14] The optimal position of the ether oxygen in the chain of Salmeterol for both potency and duration is six carbon atoms from the basic nitrogen.[18] |
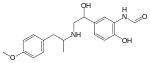 | Formoterol | Has an N-isopropyl-p-methoxyphenyl group that gives direct-action to β-receptors. Formeterol also has a m-formamide and p—hydroxyphenyl ring, which gives selectivity to β2-receptors. Formeterol’s onset of action is about 20 minutes, but duration of action of 12 hours. If Formeterol is compared to Salmeterol then it has greater water solubility that allows it to get to receptor sites faster and the lipophilicity keeps it in the lungs longer. Formeterol has two asymmetric centers. The R,R-enantiomer is reported to be 1000 times more active then S,S-enantiomer.[14] |
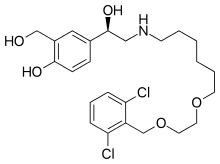 | Vilanterol | The latest drug on the market. It has similar structure to Salmeterol but also 2,6 dichlorobenzyl and two alkoxy groups on the N-chain. It has demonstrated similar selectivity as salmeterol for β2-receptor but greater selectivity compared to all other agonists tested. The intrinsic activity was demonstrated to be greater than salmeterol.[19] Vilanterol is very potent and has a high potency. It has a fast onset of action and long duration of action. The inclusion of the extra alkoxy group on the side chain gives a potent β2-agonist that is rapidly metabolized in human liver. The 2,6-dichlorobenzyl seems to give greater potency, selectivity, rapid onset of action, long duration of action and rapid turnover.[18] |
Synthesis of β2-adrenoceptor agonists
The β2-agonsist that are clinically used are all substituted β-phenethylamine (see figure 5) and they have three kinds of phenyl rings shown in figure 4. They are called resorcinol ring, salicyl alcohol ring or N-formamide ring. The alcohol substituents in the phenyl ring are reactive and complicate the synthesis of the β2-agonists. A protection step is needed while the N-residue is added in position R1 (figure 5). Another thing that complicates the synthesis is obtaining optically pure R (-) enantiomer of the compound. The stereochemistry is very important for activity because only the R (-) enantiomer is able to form the hydrogen bonds necessary to fit in the binding site and activate the β2-receptor.[14]
Salbutamol is usually inhaled in racemic mixtures (for example Ventolin). By treating asthma with optically pure (R)-salbutamol the risk of side effects, such as nervous system stimulatory effects and cardiac arrythmia, can be minimized.[20] This is why several ways to obtain optically pure salbutamol have been described but they have not been potent enough to use in the pharmaceutical industry. The most effective way to obtain pure (R)-salbutamol is to produce a racemic mixture and then separate the isomers.[21]
Stereoselective synthesis of tributalin and salbutamol acetal can be done from O-protected-(R)-cyanohydrins. F. Effenberg, et al. describe a way for the synthesis. The main complications are to perform the deprotection step without racemization and to form a pure salt. A Ritter reaction can be used for an N-tertiary butylation. In this experiment deacetylation of (R)-salbutamol acetal was unsuccessful so it can not be used to obtain pure (R)-salbutamol. Figure 6 shows the main steps in the synthesis of salmeterol and tributalin.[22]
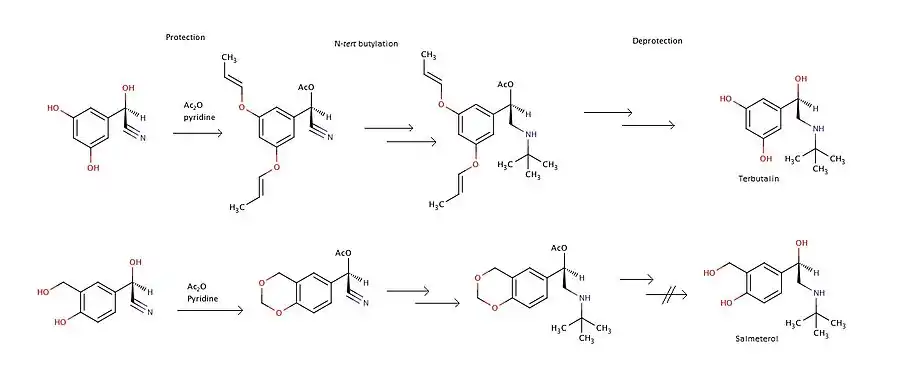
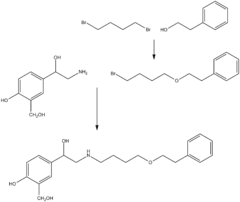
The long acting β2-agonist salmeterol can be synthesised from phenethyl alcohol. Dibromohexane is used to link the phenethyl alcohol with the salicyl alcohol ethylamine. Figure 7 shows the main steps for the synthesis.[23]
See also
References
- Tattersfield, A.E. (2006). "Current issues with beta2-adrenoceptor agonists: historical background". Am J Med. 68 (4): 471–472. doi:10.1385/CRIAI:31:2:107. PMID 17085787.
- Donohue, J. F.; Niewoehner, D.; Brooks, J.; O'Dell, D.; Church, A. (2014). "Safety and tolerability of once-daily umeclidinium/vilanterol 125/25 mcg and umeclidinium 125 mcg in patients with chronic obstructive pulmonary disease: results from a 52-week, randomized, double-blind, placebo-controlled study". Respir Res. 15: 78. doi:10.1186/1465-9921-15-78. PMC 4113670. PMID 25015176.
- "What Is Asthma?". nhlbi.nih.gov. Retrieved 22 October 2014.
- Celli, B.R.; Macnee, W. (2006). "Standards for the diagnosis and treatment of patients with COPD: A summary of the ATS/ERS position paper". European Respiratory Journal. 27 (1): 242. doi:10.1183/09031936.06.00129305.
- Barner, P.J. (2002). "Scientific rationale for inhaled combination therapy with long-acting beta2-agonists and corticosteroids". Eur Respir J. 19 (1): 182–191. doi:10.1183/09031936.02.00283202.
- Homer, C.J. (1997). "Asthma disease management". N Engl J Med. 337 (20): 1461–1463. doi:10.1056/nejm199711133372010. PMID 9358146.
- Lazarinis, N; Jorgensen, L; Ekstrom, T; Bjermer, L; Dahlen, B; Pulleritis, T; Larsson, K (2014). ". Combination of budesonide/formoterol on demand improves asthma control by reducing exercise-induced bronchoconstriction". Thorax. 69 (2): 130–136. doi:10.1136/thoraxjnl-2013-203557. PMC 3913208. PMID 24092567.
- McFadden, E.R Jr. (1980). "Exercise-induced asthma". Am J Med. 68 (4): 471–472. doi:10.1016/0002-9343(80)90282-x.
- Boulet, L.P (1994). "Long- Versus Short-Acting β2-Agonists". Drugs. 47 (2): 207–222. doi:10.2165/00003495-199447020-00001. PMID 7512898.
- "asthma care quick reference, diagnosing and managing asthma" (PDF). nhlbi.nih.gov. National Heart, Lung, and Blood Institute. p. 4. Retrieved 24 October 2014.
- Johnson, M (2006). "Molecular mechanisms of beta(2)-adrenergic receptor function, response, and regulation". J Allergy Clin Immunol. 117 (1): 18–24. doi:10.1016/j.jaci.2005.11.012. PMID 16387578.
- Armstrong, D. J.; Mottram, D. R. (2010). "Beta-2 agonists". Drugs in Sport. 98.
- Hochhaus, G; Mollmann, H (1992). "Pharmacokinetic Pharmacodynamic Characteristics of the Beta-2-Agonists Terbutaline, Salbutamol and Fenoterol". International Journal of Clinical Pharmacology and Therapeutics. 30 (9): 342–362.
- Lemke, T.L.; Williams, D.A.; Roche, V.F.; Zito, S.W. (2013). Foye's principles of medicinal chemistry. Philadelphia, PA: Lippincott Williams & Wilkins. pp. 1314–1320.
- Ladage, R. H. G; Schwinger, D; Brixius, K (2013). "Cardio-Selective Beta-Blocker: Pharmacological Evidence and Their Influence on Exercise Capacity". Cardiovascular Therapeutics. 31 (2): 76–83. doi:10.1111/J.1755-5922.2011.00306.X. PMID 22279967.
- King, F. D. (2002). Medicinal Chemistry Principles and Practice (2nd ed.). Cambridge, UK: The Royal Society of Chemistry. pp. 1ö24.
- Anderson, G. P.; Linden, A.; Rabe, K. F. (1994). "Why are long-acting beta-adrenoceptor agonists long-acting?". Eur Respir J. 7 (3): 569–578. doi:10.1183/09031936.94.07030569. PMID 7912202.
- Procopiou, Panayiotis A.; Barrett, Victoria J.; Bevan, Nicola J.; Biggadike, Keith; Box, Philip C.; Butchers, Peter R.; Coe, Diane M.; Conroy, Richard; Emmons, Amanda; Ford, Alison J.; Holmes, Duncan S.; Horsley, Helen; Kerr, Fern; Li-Kwai-Cheung, Anne-Marie; Looker, Brian E.; Mann, Inderjit S.; McLay, Iain M.; Morrison, Valerie S.; Mutch, Peter J.; Smith, Claire E.; Tomlin, Paula (2010). "Synthesis and Structure−Activity Relationships of Long-acting β Adrenergic Receptor Agonists Incorporating Metabolic Inactivation: An Antedrug Approach". Journal of Medicinal Chemistry. 53 (11): 4522–4530. doi:10.1021/jm100326d. PMID 20462258.
- Slack, R.J.; Barret, V.J.; Morrison, V.S.; Sturton, R.G.; Emmons, A.J.; Ford, A.J.; Knowles, R.G. (2013). "In Vitro Pharmacological Characterization of Vilanterol, a Novel Long-Acting β2-Adrenoceptor Agonist with 24-Hour Duration of Action". The Journal of Pharmacology and Experimental Therapeutics. 344 (1): 218–230. doi:10.1124/jpet.112.198481. PMID 23131596.
- Barberich, T.J. "Method for treating asthma using optically pure (R)-albuterol". Google Patents. Retrieved 27 October 2014.
- Hettich, Jerome. "The Chemistry, Biochemistry and Applications of Salbutamol". Imperial College - Department of Chemistry. Retrieved 27 October 2014.
- Effenberger, Franz; Jäger, Jürgen (1997). "Synthesis of the Adrenergic Bronchodilators (R)-Terbutaline and (R)-Salbutamol from (R)-Cyanohydrins". J. Org. Chem. 62 (12): 3867–3873. doi:10.1021/jo970032d.
- Skidmore, I. F.; Lunts, L. H. C.; Finch, H.; Naylor, A.; German Offen., 1984, 3414752; Chem. Abstr., 1986, 102, 95383.
