Discovery and development of statins
The discovery of HMG-CoA (3-hydroxy-3-methylglutaryl-CoA) reductase inhibitors, called statins, was a breakthrough in the prevention of hypercholesterolemia and related diseases. Hypercholesterolemia is considered to be one of the major risk factors for atherosclerosis which often leads to cardiovascular, cerebrovascular and peripheral vascular diseases.[1] The statins inhibit cholesterol synthesis in the body and that leads to reduction in blood cholesterol levels, which is thought to reduce the risk of atherosclerosis and diseases caused by it.[2]
History
In the mid-19th century, a German pathologist named Rudolf Virchow discovered that cholesterol was to be found in the artery walls of people that died from occlusive vascular diseases, like myocardial infarction. The cholesterol was found to be responsible for the thickening of the arterial walls and thus decreasing the radius in the arteries which leads in most cases to hypertension and increased risk of occlusive vascular diseases.[2]
In the 1950s the Framingham heart study led by Dawber revealed the correlation between high blood cholesterol levels and coronary heart diseases. Following up from that study the researchers explored a novel way to lower blood cholesterol levels without modifying the diet and lifestyle of subjects suffering with elevated blood cholesterol levels. The primary goal was to inhibit the cholesterol biosynthesis in the body. Hence HMG-CoA reductase (HMGR) became a natural target. HMGR was found to be the rate-limiting enzyme in the cholesterol biosynthetic pathway. There is no build-up of potentially toxic precursors when HMGR is inhibited, because hydroxymethylglutarate is water-soluble and there are alternative metabolic pathways for its breakdown.[2][3]
In the 1970s the Japanese microbiologist Akira Endo first discovered natural products with a powerful inhibitory effect on HMGR in a fermentation broth of Penicillium citrinum, during his search for antimicrobial agents. The first product was named compactin (ML236B or mevastatin). Animal trials showed very good inhibitory effect as in clinical trials, however in a long term toxicity study in dogs it resulted in toxic effects at higher doses and as a result was believed to be too toxic to be given to humans. In 1978, Alfred Alberts and colleagues at Merck Research Laboratories discovered a new natural product in a fermentation broth of Aspergillus terreus, their product showed good HMGR inhibition and they named the product mevinolin, which later became known as lovastatin.[2][3][4]
The cholesterol controversy began in the early promotion of statins.[2]
Mechanism
Statins are a competitive antagonists of HMG CoA, as they directly compete with the endogenous substrate for the active site cavity of HMGR. Statins are also noncompetitive with the cosubstrate NADPH (nicotinamide adenine dinucleotide phosphate).[5] By blocking the HMGR enzyme they inhibit the synthesis of cholesterol via the mevalonate pathway. The end result is lower LDL (Low Density Lipoprotein), TG (Triglycerides) and total cholesterol levels as well as increased HDL (High Density Lipoprotein) levels in serum.[2][3][4]
Interactive pathway map
Click on genes, proteins and metabolites below to link to respective articles. [§ 1]
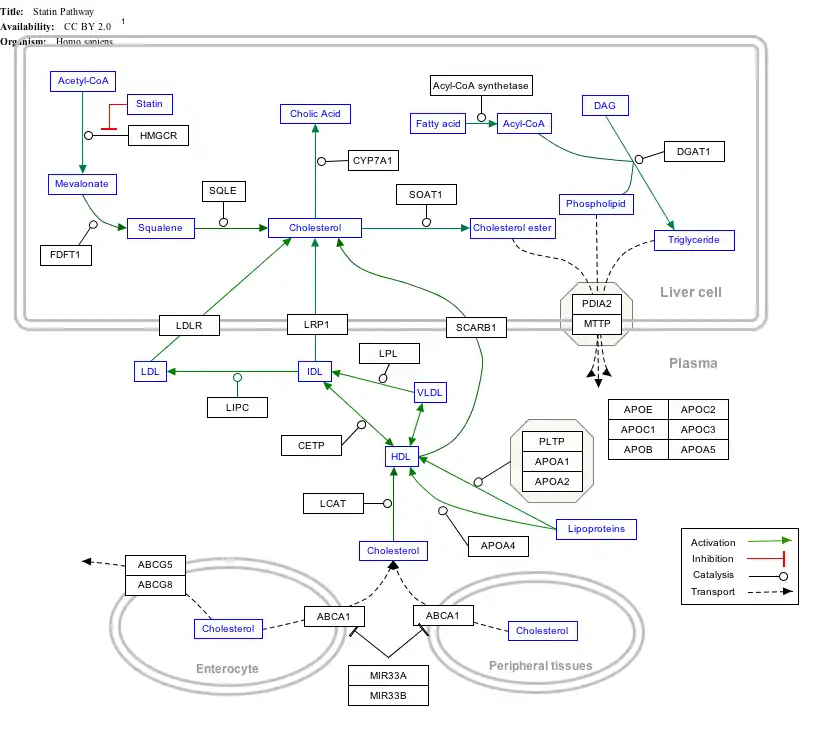
- The interactive pathway map can be edited at WikiPathways: "Statin_Pathway_WP430".
Statin drug design
The ideal statin should have the following properties:[6]
- High affinity for the enzyme active site
- Marked selectivity of uptake into hepatic cells compared with non-hepatic cells
- Low systemic availability of active inhibitory equivalents
- Relatively prolonged duration of effect.
One of the main design objectives of statin design is the selective inhibition of HMGR in the liver, as cholesterol synthesis in non-hepatic cells is needed for normal cell function and inhibition in non-hepatic cells could possibly be harmful.[7]
The statin pharmacophore
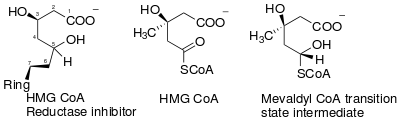
The essential structural components of all statins are a dihydroxyheptanoic acid unit and a ring system with different substituents. The statin pharmacophore is modified hydroxyglutaric acid component, which is structurally similar to the endogenous substrate HMG CoA and the mevaldyl CoA transition state intermediate (Figure 1). The statin pharmacophore binds to the same active site as the substrate HMG-CoA and inhibits the HMGR enzyme. It has also been shown that the HMGR is stereoselective and as a result all statins need to have the required 3R,5R stereochemistry.[8]
Differences in statin structure
The statins differ with respect to their ring structure and substituents. These differences in structure affect the pharmacological properties of the statins, such as:[6]
- Affinity for the active site of the HMGR
- Rates of entry into hepatic and non-hepatic tissues
- Availability in the systemic circulation for uptake into non-hepatic tissues
- Routes and modes of metabolic transformation and elimination
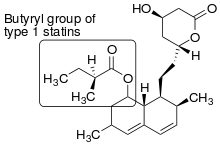
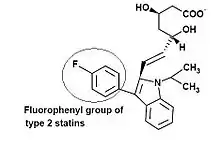
Statins have sometimes been grouped into two groups of statins according to their structure.[9]
Type 1 statins Statins that have substituted decalin-ring structure that resemble the first statin ever discovered, mevastatin have often been classified as type 1 statins due to their structural relationship. Statins that belong to this group are:[9]
- Lovastatin (Figure 2)
- Pravastatin
- Simvastatin
Type 2 statins Statins that are fully synthetic and have larger groups linked to the HMG-like moiety are often referred to as type 2 statins. One of the main differences between the type 1 and type 2 statins is the replacement of the butyryl group of type 1 statins by the fluorophenyl group of type 2 statins. This group is responsible for additional polar interactions that causes tighter binding to the HMGR enzyme. Statins that belong to this group are:[9]
- Fluvastatin (Figure 3)
- Cerivastatin
- Atorvastatin
- Rosuvastatin
Lovastatin is derived from a fungus source and simvastatin and pravastatin are chemical modifications of lovastatin and as a result do not differ much in structure from lovastatin.[7] All three are partially reduced napthylene ring structures. Simvastatin and lovastatin are inactive lactones which must be metabolized to their active hydroxy-acid forms in order to inhibit HMGR.[7] Type 2 statins all exist in their active hydroxy-acid forms. Fluvastatin has indole ring structure, while atorvastatin and rosuvastatin have pyrrole and pyrimidine based ring structure respectively. The lipophilic cerivastatin has a pyridine-based ring structure.
HMGR statin binding site
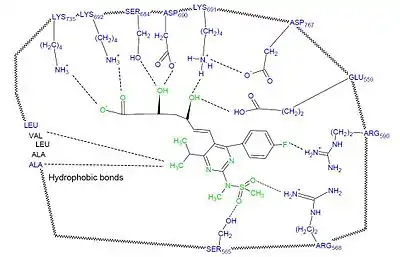
Studies have shown that statins bind reversibly to the HGMR enzyme. The affinity of statins for HGMR enzyme is in the nanomolar range, while the natural substrate's affinity is in the micromolar range.[10] Studies have shown that statins use the conformational flexibility of the HMGR enzyme that causes a shallow hydrophobic groove that the statins exploit and is used to accommodate their hydrophobic moieties.[11] The specificity and the tight binding of statins is due to orientation and bonding interactions that form between the statin and the HMGR enzyme.[9] Polar interactions are formed between the HMG-moiety and residues that are located in the cis loop of the enzyme. These polar interactions are between Ser684, Asp690, Lys691 and Lys692 (Figure 4). The terminal carboxylate of the HMG moiety forms a salt bridge with the cationic Lys735 of the enzyme. In addition to the polar interaction, Lys691 participates in a hydrogen bonding network with Glu559, Asp767 and the O5 hydroxyl group of the hydroxyglutartic acid component of the statins. Van der Waals interactions are formed between the hydrophobic side chains of the enzyme, which involve the Leu562, Val683, Leu853, Ala856 and Leu857 and the statins.[9] Type 2 statins form polar interaction between the fluorine atom on the fluorophenyl group and the guanidinium group of Arg590.[11] In addition to these interactions atorvastatin and rosuvastatin also form unique hydrogen bonds between Ser565 residue and either a carbonyl oxygen atom (atorvastatin) or a sulfone oxygen atom (rosuvastatin). A unique polar interaction between the Arg568 side chain and the electronegative sulfone group on rosuvastatin makes it the statin that has the greatest number of bonding interactions with HGMR.[9]
Structure-activity relationship (SAR)
All statins have the same pharmacophore so the difference in their pharmacodynamic effect is mostly based on the substituents. The activity of each statin is dependent on the binding affinity of the compound for the substrate site and the length of time it binds to the site.[5] Type 2 statins have unique fluorophenyl group that causes additional polar interaction between the enzyme and the statins, which results in a tighter binding to the enzyme. The newest statin, rosuvastatin has a unique polar methane sulfonamide group, which is quite hydrophilic and confers low lipophilicity. The sulfonamide group forms a unique polar interaction with the enzyme. As a result, rosuvastatin has superior binding affinity to the HMGR enzyme compared to the other statins, which is directly related to its efficiency to lower LDL cholesterol.[6]
Lipophilicity
Lipophilicity of the statins is considered to be quite important since the hepatoselectivity of the statins is related to their lipophilicity. The more lipophilic statins tend to achieve higher levels of exposure in non-hepatic tissues, while the hydrophilic statins tend to be more hepatoselective. The difference in selectivity is because lipophilic statins passively and non-selectively diffuse into both hepatocyte and non-heptatocyte, while the hydrophilic statins rely largely on active transport into hepatocyte to exert their effects.[5][12] High hepatoselectivity is thought to translate into reduced risk of adverse effects.[7] It has been reported that the organic anion transporting polypeptide (OATP) is important for the hepatic uptake of hydrophilic statins such as rosuvastatin and pravastatin.[5][12] OATP-C is expressed in liver tissue on the basolateral membrane of hepatocytes and is considered to be a potential contributor for the low IC50 for rosuvastatin in hepatocytes. Of the marketed statins, cerivastatin was the most lipophilic and also had the largest percentage of serious adverse effects due to its ability to inhibit vascular smooth muscle proliferation and as a result was voluntarily removed from the market by the manufacturer.[5]
| Cerivastatin | Simvastatin | Fluvastatin | Atorvastatin | Rosuvastatin | Pravastatin | |
|---|---|---|---|---|---|---|
| Log D Class | 1,50–1,75 | 1,50–1,75 | 1,00–1,25 | 1,00–1,25 | -0,25–(-0,50) | -0,75–(-1,0) |
Metabolism
All statins are (metabolized) by the liver, which causes their low systemic bioavailability.[13] Lovastatin and simvastatin are administered in their lactone forms, which is more lipophilic than their free acid forms, and therefore they have to be activated by hydrolysis to the active anionic carboxylate form.[8][13] Cytochrome P450(CYP) isoenzymes are involved in the oxidative metabolism of the statins, with CYP3A4 and CYP2C9 isoenzymes being the most dominant. CYP3A4 isoenzyme is the most predominant isoform involved in metabolism of lovastatin, simvastatin, atorvastatin and cerivastatin.[8][13] CYP2C9 isoenzyme is the most predominant isoform involved in metabolism of Fluvastatin, but CYP3A4 and CYP2C8 isoenzymes also contribute to the metabolism of Fluvastatin.[13] Rosuvastatin is metabolized to a small degree by CYP2C9 and to a lesser extent by CYP2C19 isoenzymes. Pravastatin is not metabolized by CYP isoenzymes to any appreciable extent.[6][8][13] The statins that have the ability to be metabolized by multiple CYP isoenzymes may therefore avoid drug accumulation when one of the pathways is inhibited by co-administered drugs.[13]
Comparative pharmacology of statins
| Drug | Reduction in LDL-C (%) | Increase in HDL-C (%) | Reduction in TG (%) | Reduction in TC (%) | Metabolism | Protein binding (%) | T1/2 (h) | Hydrophilic | IC50 (nM)[6] |
|---|---|---|---|---|---|---|---|---|---|
| Atorvastatin | 26 – 60 | 5 – 13 | 17 – 53 | 25 – 45 | CYP3A4 | 98 | 13–30 | No | 8 |
| Lovastatin | 21 – 42 | 2 – 10 | 6 – 27 | 16 – 34 | CYP3A4 | >95 | 2 – 4 | No | NA |
| Simvastatin | 26 – 47 | 8 – 16 | 12 – 34 | 19 – 36 | CYP3A4 | 95 – 98 | 1 – 3 | No | 11 |
| Fluvastatin | 22 – 36 | 3 – 11 | 12 – 25 | 16 – 27 | CYP2C9 | 98 | 0,5 – 3,0 | No | 28 |
| Rosuvastatin | 45 – 63 | 8 – 14 | 10 – 35 | 33 – 46 | CYP2C9 | 88 | 19 | Yes | 5 |
| Pravastatin | 22 – 34 | 2 – 12 | 15 – 24 | 16 – 25 | Sulfation | 43 – 67 | 2 – 3 | Yes | 44 |
Future research
With the recent elucidation of the structures of the catalytic portion of human HMGR enzyme complexed with six different statins by a series of crystallography studies, new possibilities have opened up for the rational design and optimization of even better HGMR inhibitors.[15]
A new study using comparative molecular field analysis (CoMFA) to establish three-dimensional quantitative structure-activity relationship (3D QSAR), while searching for novel active pharmacophores as potentially potent HGMR inhibitors, was recently published. Using this novel technique researchers were able to screen for compounds with high screening scores. In addition to the conventional statin-like compounds with HMG-like moiety, eight additional compounds with completely different pharmacophore structure were found. This structure-based virtual screening procedure is considered promising for rational quest and optimization of potential novel HGMR inhibitors.[15]
References
- Christians, Uwe; Jacobsen, Wolfgang; Floren, Leslie C. (October 1998). "Metabolism and Drug Interactions of 3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase Inhibitors in Transplant Patients: Are the Statins Mechanistically Similar?". Pharmacology and Therapeutics. 80 (1): 1–34. doi:10.1016/S0163-7258(98)00016-3. PMID 9804052.
- Tobert, Jonathan A. (July 2003). "Lovastatin and beyond: The history of the HMG-CoA reductase inhibitors". Nature Reviews Drug Discovery. 2 (7): 517–526. doi:10.1038/nrd1112. PMID 12815379.
- Endo, Akira (November 1, 1992). "The Discovery and development of HMG-CoA reductase inhibitors". Journal of Lipid Research. 33 (11): 1569–80. PMID 1464741.
- Endo, Akira (2004). "The origin of the statins". International Congress Series. 1262: 3–8. doi:10.1016/j.ics.2003.12.099.
- White, C. Michael (September 1, 2002). "A review of the pharmacologic and pharmacokinetic aspects of rosuvastatin". The Journal of Clinical Pharmacology. 42 (9): 963–970. doi:10.1177/009127002401102876. PMID 12211221. Archived from the original on March 21, 2008. Retrieved November 9, 2007.
- McTaggart, Fergus (2003). "Comparative pharmacology of rosuvastatin". Atherosclerosis Supplements. 4 (1): 9–14. doi:10.1016/S1567-5688(03)00004-7. PMID 12714032.
- Hamelin, Bettina A.; Turgeon, Jacques (January 1998). "Hydrophilicity/lipophilicity: relevance for the pharmacology and clinical effects of HMG-CoA reductase inhibitors". Trends in Pharmacological Sciences. 19 (1): 26–37. doi:10.1016/S0165-6147(97)01147-4. PMID 9509899.
- Roche, Victoria F. (2005). "Teachers' Topics: Antihyperlipidemic Statins: A Self-Contained, Clinically Relevant Medicinal Chemistry Lesson" (PDF). American Journal of Pharmaceutical Education. 69 (4): 546–560. doi:10.5688/aj690477. Archived from the original (PDF) on 2007-10-28.
- Istvan, Eva S.; Deisenhofer, Johann (May 2001). "Structural Mechanism for Statin Inhibition of HMG-CoA Reductase". Science Magazine. 292 (5519): 1160–4. doi:10.1126/science.1059344. PMID 11349148.
- Moghadasian, Mohammed H. (May 1999). "Clinical pharmacology of 3-hydroxy-methylglutaryl coenzyme A reductase inhibitors". Life Sciences. 65 (13): 1329–37. doi:10.1016/S0024-3205(99)00199-X. PMID 10503952.
- Istvan, Eva S. (December 2002). "Structural mechanism for statin inhibition of 3-hydroxy-3methylglutaryl coenzyme A reductase". American Heart Journal. 144 (6): 27–32. doi:10.1067/mhj.2002.130300. PMID 12486413.
- Pfefferkorn, Jeffrey A.; Song, Yuntao; Sun, Kuai-Lin; Miller, Steven R.; Trivedi, Bharat K.; Choi, Chulho; Sorensen, Roderick J.; Bratton, Larry D.; Unangst, Paul C. (June 2007). "Design and synthesis of hepatoselective, pyrrole-based HMG-CoA reductase inhibitors". Bioorganic & Medicinal Chemistry Letters. 17 (16): 4538–44. doi:10.1016/j.bmcl.2007.05.096. PMID 17574412.
- Corsini, Alberto; Bellosta, Stefano; Baetta, Roberta; Fumagalli, Remo; Paoletti, Rodolfo; Bernini, Franco (1999). "New insights into the pharmacodynamic and pharmacokinetic properties of statins". Pharmacology & Therapeutics. 84 (3): 413–428. doi:10.1016/S0163-7258(99)00045-5. PMID 10665838.
- Vaughan, Carl J.; Gotto, Jr., Antonio M. (June 2004). "Update on Statins: 2003". Circulation. 110 (7): 886–892. doi:10.1161/01.CIR.0000139312.10076.BA. PMID 15313959.
- Zhang, Qing Y.; Wan, Jian; Xu, Xin; Yang, Guang F.; Ren, Yan L.; Liu, Jun J.; Wang, Hui; Guo, Yu (November 2006). "Structure-Based Rational Quest for Potential Novel Inhibitors of Human HMG-CoA Reductase by Combining CoMFA 3D QSAR Modeling and Virtual Screening". Journal of Combinatorial Chemistry. 9 (1): 131–8. doi:10.1021/cc060101e. PMID 17206841.