Klippel–Feil syndrome
Klippel–Feil syndrome (KFS), also known as cervical vertebral fusion syndrome, is a rare congenital condition characterized by the abnormal fusion of any two of the seven bones in the neck (cervical vertebrae).[1]:578 It results in a limited ability to move the neck and shortness of the neck, resulting in the appearance of a low hairline.[2]
| Klippel-Feil syndrome | |
|---|---|
| Other names | Congenital dystrophia brevicollis, cervical vertebral fusion syndrome |
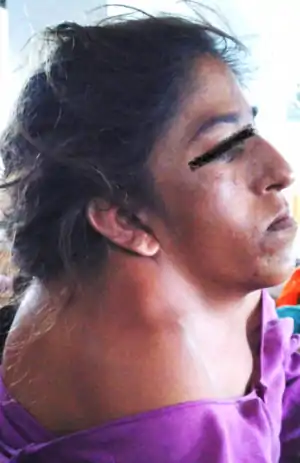 | |
| Woman with Klippel–Feil syndrome | |
| Pronunciation |
|
| Specialty | Paediatrics, orthopaedics |
| Symptoms | Cervical spine fusion, scoliosis, spina bifida, heart defect, respiratory problems, other syndromic features |
| Usual onset | Congenital |
| Causes | Genetic mutations |
| Risk factors | Family history |
| Prognosis | Shorter life expectancy in some cases |
| Frequency | 1 in 40,000 to 42,000 births, females more affected than males |
The syndrome is difficult to diagnose, as it occurs in a group of patients affected with many different abnormalities who can only be unified by the presence of fused or segmental cervical vertebrae.[3] KFS is not always genetic and not always known the day of the birth.
The disease was initially reported in 1884 by Maurice Klippel and André Feil from France.[4] In 1919, in his Doctor of Philosophy thesis,[5] André Feil suggested another classification of the syndrome, encompassing not only deformation of the cervical spine, but also deformation of the lumbar and thoracic spine.
Signs and symptoms
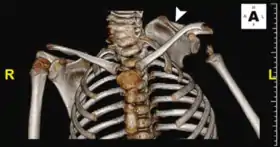
KFS is associated with many other abnormalities of the body, hence thorough evaluation of all patients with fused cervical vertebrae at birth is required. Furthermore, it is unclear whether KFS is a unique disease, or if it is one part of a spectrum of congenital spinal deformities.
KFS is usually diagnosed after birth. The most common signs of the disorder are restricted mobility of the neck and upper spine and a shortened neck with the appearance of a low hairline at the back of the head.
Associated abnormalities may include:[6][7][8][9]
- Scoliosis (sideways curvature of the spine)
- Spina bifida
- Problems with the kidneys and the ribs
- Cleft palate
- Dental problems (delayed dentition, cavities, missing teeth)
- Respiratory problems
- Heart defects
- Short stature
- Duane syndrome
- Srb's anomaly
- Sprengel's deformity
The disorder also may be associated with abnormalities of the head and face, skeleton, sex organs, muscles, brain and spinal cord, arms, legs and fingers.
Genetics
Mutations of the GDF6, GDF3 and MEOX1 gene are associated with KFS.[2] The cause of the condition is unknown in individuals with KFS who do not have mutations of these two genes. GDF6 and GDF3 provide the body with instructions for making proteins involved in regulating the growth and maturation of bone and cartilage. GDF6 specifically is involved in the formation of vertebral bones, among others, and establishing boundaries between bones in skeletal development. GDF3 is involved with bone and cartilage growth. Mutations of GDF6, GDF3 and MEOX1 cause a reduced number of functional proteins that are coded by these genes, but it is unclear exactly how a shortage in these proteins leads to incomplete separation of the vertebrae in people with KFS.[10] However, when the GDF6 gene was removed in mice, the result was the fusion of bones.[11]
These mutations can be inherited in two ways:
- Autosomal dominant inheritance, where one copy of the altered gene in each cell is sufficient to cause the disorder, is especially associated with C2-C3 fusion.[10]
- Autosomal recessive inheritance, where both copies of a gene contain mutations, is especially associated with C5-C6 fusion.[10]
- Another autosomal dominant form (mapped on locus 8q22.2), known as KFS with laryngeal malformation, has been identified. It is also known as segmentation syndrome 1.[12][13]
Diagnosis
The heterogeneity of KFS has made it difficult to outline the diagnosis as well as the prognosis for this disease.
Classification
In 1912, Maurice Klippel and Andre Feil independently provided the first descriptions of KFS. They described patients who had a short, webbed neck; decreased range of motion (ROM) in the cervical spine; and a low hairline. Feil subsequently classified the syndrome into 3 categories:
- Type I — Fusion of C2 and C3 with occipitalization of the atlas. In 1953, further complications were later reported by McRae; flexion and extension is concentrated within the C1 and C2 vertebrae. As with aging, the odontoid process can become hypermobile, narrowing the space where the spinal cord and brain stem travel (spinal stenosis).
- Type II — Long fusion below C2 with an abnormal occipital-cervical junction. Similar to the C2-C3 fusion of McRae and could be viewed as a more elaborate variation. Flexion, extension, and rotation are all concentrated in the area of an abnormal odontoid process or poorly developed ring of C1 which cannot withstand the effects of aging.
- Type III — A single open interspace between two fused segments. Cervical spine motion is concentrated at single open articulation. This hypermobility may lead to instability or degenerative osteoarthritis. This pattern can be recognized as the cervical spine is often seen to be at an angle or hinge at this open segment.
A classification scheme for KFS was proposed in 1919 by Andre Feil, which accounted for cervical, thoracic, and lumbar spine malformations.[14]
However, in 2006, Dino Samartzis and colleagues proposed three classification-types that specifically addressed the cervical spine anomalies and their associated cervical spine-related symptoms, with additional elaboration on various time-dependent factors regarding this syndrome.[15]
Treatment
Treatment for KFS is symptomatic and may include surgery to relieve cervical or craniocervical instability and constriction of the spinal cord, and to correct scoliosis.
If symptomatic treatment fails, spinal surgery may provide relief. Adjacent segment disease and scoliosis are two examples of common symptoms associated with Klippel–Feil syndrome, and they may be treated surgically. The three categories treated for types of spinal cord deficiencies are massive fusion of the cervical spine (Type I), the fusion of 1 or 2 vertebrae (Type II), and the presence of thoracic and lumbar spine anomalies in association with type I or type II Klippel–Feil syndrome (Type III).
Adjacent segment disease can be addressed by performing cervical disc arthroplasty using a device such as the Bryan cervical disc prosthesis.[16] The option of the surgery is to maintain range of motion and attenuate the rate of adjacent segment disease advancement without fusion.[17] Another type of arthroplasty that is becoming an alternate choice to spinal fusion is Total Disc Replacement. Total disc replacement objective is to reduce pain or eradicate it.[18] Spinal fusion is commonly used to correct spinal deformities such as scoliosis. Arthrodesis is the last resort in pain relieving procedures, usually when arthroplasties fail.
Prognosis
The prognosis for most individuals with KFS is good if the disorder is treated early and appropriately. Activities that can injure the neck should be avoided, as it may contribute to further damage. Other diseases associated with the syndrome can be fatal if not treated, or if found too late to be treatable.[19]
In less than 30% of cases, individuals with KFS will present with heart defects.[20] If these heart defects are present, they often lead to a shortened life expectancy, the average being 35–45 years of age among males and 40–50 among females. This condition is similar to the heart failure seen in gigantism.[21]
Epidemiology
The prevalence of KFS is unknown due to the lack of studies to determine its prevalence.[22] It is estimated to occur 1 in 40,000 to 42,000 newborns worldwide. In addition, females seem to be affected slightly more often than males.[10]
Notable cases
Ancient
- A case of a child in Switzerland was discovered in a necropolis dated between 4500 and 4000 BC.[23]
- In 2009, archaeologists excavating at a Neolithic site of the Đa Bút culture of northern Vietnam discovered the remains of a young man around age 25, "Burial 9", living between 2000 BC and 1500 BC with Klippel–Feil syndrome, who had apparently been supported by his subsistence-level community for at least a decade before his death.[24][25][26]
- The 18th Dynasty Egyptian pharaoh Tutankhamun is believed by some to have suffered from Klippel–Feil syndrome,[27] though others dispute this claim.[28]
Contemporary
- English cricketer Gladstone Small[29]
- "Big" Ed Brown from the TLC series, 90 Day Fiancé.
References
- Andrews, James, William; Berger, Timothy; Elston, Dirk (2005). Andrews' Diseases of the Skin: Clinical Dermatology. (10th ed.). Saunders. ISBN 0-7216-2921-0.
- "Klippel-Feil syndrome". Genetics Home Reference. U.S. National Library of Medicine. Retrieved 2018-08-18.
- "Klippel-Feil Syndrome". HONselect.
- Belykh, Evgenii; Malik, Kashif; Simoneau, Isabelle; Yagmurlu, Kaan; Lei, Ting; Cavalcanti, Daniel D.; Byvaltsev, Vadim A.; Theodore, Nicholas; Preul, Mark C. (July 2016). "Monsters and the case of L. Joseph: André Feil's thesis on the origin of the Klippel-Feil syndrome and a social transformation of medicine". Neurosurgical Focus. 41 (1): E3. doi:10.3171/2016.3.FOCUS15488. ISSN 1092-0684. PMID 27364256.
- Feil A (1919). "These de medicine, Paris. L'absence et la diminution des vertèbres cervicales (étude clinique et pathogénique); le syndrome de réduction numérique cervicales". Cite journal requires
|journal=(help) - Paradowska, Szeląg, Sławecki (2007). "Klippel−Feil Syndrome – Review of the Literature" (PDF). Dent. Med. Probl. Archived from the original (PDF) on 2015-10-01. Retrieved 2015-09-30.CS1 maint: multiple names: authors list (link)
- de Lima, Marina de Deus Moura; Ortega, Karem Lopez; Araújo, Luis Carlos Arias; Soares, Marcelo Melo; de Magalhães, Marina Helena Cury Gallottini (2009-12-01). "Dental team management for a patient with Klippel-Feil syndrome: case report". Special Care in Dentistry. 29 (6): 244–248. doi:10.1111/j.1754-4505.2009.00101.x. ISSN 1754-4505. PMID 19886936.
- Marchiori, Dennis (2004). Clinical Imaging - E-Book: With Skeletal, Chest and Abdomen Pattern Differentials. Elsevier Health Sciences. ISBN 978-0323071277. Retrieved 25 January 2018.
- Farsetti P, Weinstein SL, Caterini R, De Maio F, Ippolito E (May 2003). "Sprengel's deformity: long-term follow-up study of 22 cases". J Pediatr Orthop B. 12 (3): 202–10. doi:10.1097/01202412-200305000-00007. PMID 12703036. Sarwark, JF; LaBella, CR, eds. (2010). Pediatric Orthopaedics and Sports Injuries: A Quick Reference Guide. Elk Grove Village IL: American Academy of Pediatrics. pp. 231–4.
- at NLM Genetics Home Reference
- Tassabehji M, Fang ZM, Hilton EN, et al. (August 2008). "Mutations in GDF6 are associated with vertebral segmentation defects in Klippel-Feil syndrome". Hum. Mutat. 29 (8): 1017–27. doi:10.1002/humu.20741. PMID 18425797. S2CID 5276691.
- Online Mendelian Inheritance in Man (OMIM): Klippel-Feil Syndrome 1, Autosomal Dominant; KFS1 - 118100
- "Segmentation syndrome 1 | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program".
- Feil A. L'absence et la diminuaton des vertebres cervicales (etude cliniqueet pathogenique); le syndrome dereduction numerique cervicales. Theses de Paris; 1919.
- Samartzis DD, Herman J, Lubicky JP, Shen FH (2006). "Classification of congenitally fused cervical patterns in Klippel-Feil patients: epidemiology and role in the development of cervical spine-related symptoms". Spine. 31 (21): E798–804. doi:10.1097/01.brs.0000239222.36505.46. PMID 17023841. S2CID 19744236.
- Goffin J, Casey A, Kehr P, et al. (September 2002). "Preliminary clinical experience with the Bryan Cervical Disc Prosthesis". Neurosurgery. 51 (3): 840–5, discussion 845–7. doi:10.1227/00006123-200209000-00048. PMID 12188968.
- Papanastassiou ID, Baaj AA, Dakwar E, Eleraky M, Vrionis FD (March 2011). "Failure of cervical arthroplasty in a patient with adjacent segment disease associated with Klippel-Feil syndrome". Indian J Orthop. 45 (2): 174–7. doi:10.4103/0019-5413.77139. PMC 3051126. PMID 21430874.
- Phillips FM, Garfin SR (September 2005). "Cervical disc replacement". Spine. 30 (17 Suppl): S27–33. doi:10.1097/01.brs.0000175192.55139.69. PMID 16138062. S2CID 46420208.
- Cathy C. Cartwright; Donna C. Wallace (3 May 2007). Nursing care of the pediatric neurosurgery patient. pp. 205–. ISBN 978-3-540-29703-1. Retrieved 25 December 2010.
- https://rarediseases.info.nih.gov/diseases/10280/klippel-feil-syndrome
- McGaughran JM, Kuna P, Das V (October 1998). "Audiological abnormalities in the Klippel-Feil syndrome". Arch. Dis. Child. 79 (4): 352–5. doi:10.1136/adc.79.4.352. PMC 1717726. PMID 9875048.
- Angeli, E., Wagner, J., Lawrick, E., Moore, K., Anderson, M., Soderland, L., & Brizee, A. (2010, May 5). General format title. Retrieved from http://owl.english.purdue.edu/owl/resource/560/01/
- "Aspects historiques".
- "Oldest Known Paralyzed Human Discovered". Archived from the original on 2012-07-10. Retrieved 2014-01-18.CS1 maint: bot: original URL status unknown (link)
- Tilley, Lorna; Oxenham, Marc F (March 2011). "Survival against the odds: Modeling the social implications of care provision to seriously disabled individuals". International Journal of Paleopathology. 1 (1): 35–42. doi:10.1016/j.ijpp.2011.02.003. PMID 29539340.
- Gorman, James (2012-12-17). "Ancient Bones That Tell a Story of Compassion". The New York Times (17 December 2012, New York edition, D1). The New York Times. Retrieved 12 April 2015.
- Barrow, Becky (2002-09-29). "Tutankhamun shows his face 80 years after tomb is opened". The Telegraph. London. Retrieved 2007-07-12.
- Boyer RS, Rodin EA, Grey TC, Connolly RC (2003). "The skull and cervical spine radiographs of Tutankhamen: a critical appraisal". AJNR. American Journal of Neuroradiology. 24 (6): 1142–7. PMID 12812942.
- Hughes, Simon (1997-09-05). "Small gains from wealth of partners". Cricinfo. Retrieved 2007-12-13.
This article incorporates information in the public domain prepared by the National Institute of Neurological Disorders and Stroke.
External links
| Classification | |
|---|---|
| External resources |