Pyruvate dehydrogenase deficiency
Pyruvate dehydrogenase deficiency (also known as pyruvate dehydrogenase complex deficiency or PDCD) is a rare neurodegenerative disorders associated with abnormal mitochondrial metabolism. PDCD is a genetic disease resulting from mutations in one of the components of the pyruvate dehydrogenase complex (PDC).[1] The PDC is a multi-enzyme complex that plays a vital role as a key regulatory step in the central pathways of energy metabolism in the mitochondria.[2] The disorder shows heterogeneous characteristics in both clinical presentation and biochemical abnormality.[3]
| Pyruvate dehydrogenase complex deficiency | |
|---|---|
| Specialty | Endocrinology |
Biochemistry & Genetics
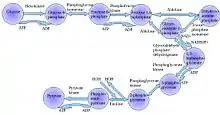
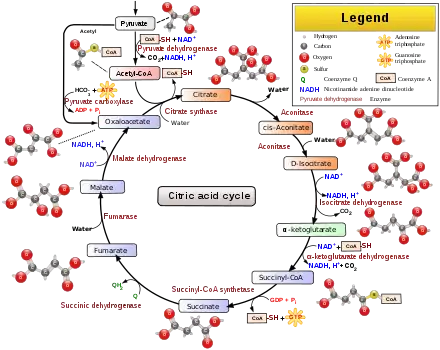
Aerobic respiration is the process of converting energy in the form of glucose into ATP, the primary currency of energy used by cells to fuel biochemical processes and support growth. The first phase of respiration is glycolysis, a series of ten biochemical reactions in the cytoplasm that convert glucose into pyruvate. Pyruvate is then transported into mitochondria, where it is converted by the pyruvate dehydrogenase complex into acetyl-CoA, the starting substrate of the Krebs cycle. When PDC activity is reduced or abolished by mutation, pyruvate levels rise. Excess pyruvate is then converted into lactic acid by lactate dehydrogenase. Lactic acid enters the blood stream, causing acidification in a condition known as lactic acidosis.
The most commonly seen form of PDCD is caused by mutations in the X-linked E1 alpha gene, PDHA1,[4] and is approximately equally prevalent in both males and females. However, males are more severely affected than heterozygous females. This can be explained by x-inactivation, as females carry one normal and one mutant gene. Cells with a normal allele active can metabolize the lactic acid that is released by the PDH deficient cells. They cannot, however, supply ATP to these cells and, therefore, phenotype depends largely on the nature/severity of the mutation.[5][6]
More rarely, mutations occur in the E2 (dihydrolipoyl transacetylase) or the E3 (dihydrolipoyl dehydrogenase) subunits of the PDC enzymatic complex, DLAT and DLD genes respectively. In these cases, PDCD displays autosomal recessive inheritance, affecting males and females equally.[7]
In cases where PDHD is a result of a mutation in a gene other than PDHA1, it is most commonly known to be due to mutations in the following four genes, PDHB, DLAT, PDHX and PDP1. All of these genes, like the PDHA1 gene are responsible for coding for a specific subunit of the pyruvate dehydrogenase complex. The PDHB gene is responsible for the coding of the E1 beta subunit of the pyruvate dehydrogenase complex. The DLAT gene is responsible for the coding of the E2 subunit, and the PDP1 is responsible for producing the PDH phosphatase catalytic subunit that catalyzes PDH dephosphorylation. This dephosphorylation activates the complex. The final gene that could be responsible for this disease is the PDHX gene, which codes for the E3 binding protein which is responsible for binding E3 dimers to the E2 subunit of the complex [8]
Epidemiology
Pyruvate dehydrogenase deficiency is extremely rare, with ~500 reported cases in the medical literature. Due to the rarity and unfamiliarity of the disease, it is likely underdiagnosed[9] (Shin et al., 2017).
Signs and symptoms
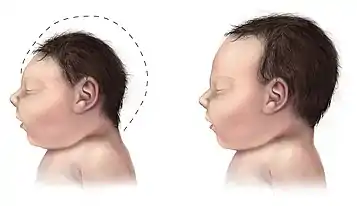
PDCD is generally presented in one of two forms. The metabolic form appears as lactic acidosis. The neurological form of PDCD contributes to hypotonia, poor feeding, lethargy and structural abnormalities in the brain.[10] Patients may develop seizures and/or neuropathological spasms. These presentations of the disease usually progress to mental retardation, microcephaly, blindness, and spasticity.[5][11][9]
Females with residual pyruvate dehydrogenase activity will have no uncontrollable systemic lactic acidosis and few, if any, neurological symptoms. Conversely, females with little to no enzyme activity will have major structural brain abnormalities and atrophy. Males with mutations that abolish, or almost abolish, enzyme activity presumably die in utero because brain cells are not able to generate enough ATP to be functionally viable. It is expected that most cases will be of mild severity and have a clinical presentation involving lactic acidosis.[6] Male infants that reach full term display more severe symptoms than females, and exhibit high mortality within the first few years of life [12][9]
Prenatal onset may present with non-specific signs such as low Apgar scores and small for gestational age. These cases display hydrocephalus, and thinning of the cerebral tissue.[9] Metabolic disturbances may also be considered with poor feeding and lethargy out of proportion to a mild viral illness, and especially after bacterial infection has been ruled out.[5] PDH activity may be enhanced by exercise, phenylbutyrate and dichloroacetate.
The clinical presentation of congenital PDH deficiency is typically characterized by heterogenous neurological features that usually appear within the first year of life. In addition, patients usually show severe hyperventillation due to profound metabolic acidosis mostly related to lactic acidosis. Metabolic acidosis in these patients is usually refractory to correction with bicarbonate.[13]
The following table lists common symptoms of pyruvate dehydrogenase deficiency.[3]
| Symptoms | Definition/Explanation |
|---|---|
| Lactic Acidosis | High levels of lactate in the blood; can cause nausea, vomiting, breathing problems, abnormal heartbeats
*In less severe cases, signs of lactic acidosis can include ataxia and episodes may only occur when ill, under stress, or after consuming high amounts of carbohydrates. |
| Hyperammonemia | High levels of ammonia in the blood; can cause confusion, weakness, fatigue |
| Facial Deformities | Narrow head, prominent forehead, wide nasal bridge, flared nostrils |
| Neurological Impairments | Developmental delays, intellectual impairments, seizures, lethargy (lack of energy), abnormal eye movements, blindness, microcephaly, poor coordination, difficulty walking |
| Abnormal Brain Structure | Underdeveloped corpus callosum, atrophy of the cerebral cortex, lesions on some parts of the brain |
| Muscular Abnormalities | Hypotonia (weak muscle tone), spasticity (tight muscles), ataxia (abnormal muscle movements) |
| Abnormalities at Infancy | Low APGAR scores (scores measuring a baby’s health after birth), low birth weight, difficulty nursing |
| Breathing Difficulties | Tachypnea (rapid breathing) |
| Fetal Abnormalities | Poor fetal weight gain, low levels of estriol in the mother’s urine |
Diagnosis

Pyruvate dehydrogenase deficiency can be diagnosed via the following methods:[14]
- Blood test (Lactate and pyruvate levels)
- Urine analysis
- Magnetic resonance spectroscopy
- MRI
Differential diagnosis
The differential diagnosis of pyruvate dehydrogenase deficiency can consist of either D-Lactic acidosis or abnormalities associated with gluconeogenesis.[14]
Treatment & Monitoring
Direct treatment that stimulates the pyruvate dehydrogenase complex (PDC), provides alternative fuels, and prevents acute worsening of the syndrome.[15] However, some correction of acidosis does not reverse all the symptoms. CNS damage is common and limits a full recovery.[9] Ketogenic diets, with high fat and low carbohydrate intake have been used to control or minimize lactic acidosis and anecdotal evidence shows successful control of the disease, slowing progress and often showing rapid improvement.[16] Ketogenic baby formulas such as Nutricia KetoCal are available.[17] With the ketogenic diet, ATP is synthesized by the catabolism of fatty acids rather than glucose, which produces the ketone bodies, 3-beta-hydroxybutyrate, acetoacetate, and acetone. Ketone bodies serve as an alternate source of energy for the body and the brain.[16] Preliminary data from PDHD patients on the ketogenic diet indicate that in milder cases, there is a reduction in the frequency of seizures, abnormal EEG readings, ataxia and abnormal sleeping patterns, and extension of remission periods. More severe cases are less responsive to the ketogenic diet, but have displayed modest improvement of gross and fine motor skills, speech and language development and development of social skills.[16] The ketogenic diet has several long term drawbacks, including pancreatitis, sialorrhea and obstipation to vomiting. Patients must be monitored regularly for blood lactate levels, transaminase and plasma ketone levels.[16]
There is some evidence that dichloroacetate reduces the inhibitory phosphorylation of pyruvate dehydrogenase complex and thereby activates any residual functioning complex. Resolution of lactic acidosis is observed in patients with E1 alpha enzyme subunit mutations that reduce enzyme stability. However, treatment with dichloroacetate does not improve neurological damage.[5] Oral citrate is often used to treat acidosis.[18]
Clinical trials to improve scientific and medical understanding of PDCD are underway. More information is located at ClinicalTrials.gov.[19]
See also
References
- Brown, G K; Otero, L J; LeGris, M; Brown, R M (November 1994). "Pyruvate dehydrogenase deficiency". Journal of Medical Genetics. 31 (11): 875–879. doi:10.1136/jmg.31.11.875. ISSN 0022-2593. PMC 1016663. PMID 7853374.
- Patel, Mulchand S.; Nemeria, Natalia S.; Furey, William; Jordan, Frank (2014-06-13). "The Pyruvate Dehydrogenase Complexes: Structure-based Function and Regulation". Journal of Biological Chemistry. 289 (24): 16615–16623. doi:10.1074/jbc.R114.563148. ISSN 0021-9258. PMC 4059105. PMID 24798336.
- Reference, Genetics Home. "pyruvate dehydrogenase deficiency". Genetics Home Reference. Retrieved 2016-11-08.
- Imbard, A.; Boutron, A.; Vequaud, C.; Zater, M.; de Lonlay, P.; de Baulny, H. Ogier; Barnerias, C.; Miné, M.; Marsac, C.; Saudubray, J.-M.; Brivet, M. (December 2011). "Molecular characterization of 82 patients with pyruvate dehydrogenase complex deficiency. Structural implications of novel amino acid substitutions in E1 protein". Molecular Genetics and Metabolism. 104 (4): 507–516. doi:10.1016/j.ymgme.2011.08.008. ISSN 1096-7206. PMID 21914562.
- G K Brown; L J Otero; M LeGris; R M Brown (November 1994). "Pyruvate dehydrogenase deficiency". J Med Genet. 31 (11): 875–879. doi:10.1136/jmg.31.11.875. PMC 1016663. PMID 7853374.
- H H Dahl (March 1995). "Pyruvate dehydrogenase E1 alpha deficiency: males and females differ yet again". Am J Hum Genet. 56 (3): 553–557. PMC 1801181. PMID 7887408.
- Kerr, D. S.; Wexler, I. D.; Tripatara, A.; Patel, M. S. (1996), Patel, Mulchand S.; Roche, Thomas E.; Harris, Robert A. (eds.), "Human defects of the pyruvate dehydrogenase complex", Alpha-Keto Acid Dehydrogenase Complexes, MCBU Molecular and Cell Biology Updates, Birkhäuser, pp. 249–269, doi:10.1007/978-3-0348-8981-0_18, ISBN 978-3-0348-8981-0
- (GeneCards - Human Genes | Gene Database | Gene Search, n.d.)
- Shin, Ha Kyung; Grahame, George; McCandless, Shawn E.; Kerr, Douglas S.; Bedoyan, Jirair K. (2017-11-01). "Enzymatic testing sensitivity, variability and practical diagnostic algorithm for pyruvate dehydrogenase complex (PDC) deficiency". Molecular Genetics and Metabolism. 122 (3): 61–66. doi:10.1016/j.ymgme.2017.09.001. ISSN 1096-7192. PMC 5722699. PMID 28918066.
- Cassandra L. Kniffin (28 October 2014). "pyruvate dehydrogenase E1-alpha deficiency; PDHAD". Online Mendelian Inheritance in Man. Johns Hopkins University. Entry # 312170. Retrieved 2016-11-09.
- Patel, Kavi P.; O'Brien, Thomas W.; Subramony, Sankarasubramon H.; Shuster, Jonathan; Stacpoole, Peter W. (January 2012). "The Spectrum of Pyruvate Dehydrogenase Complex Deficiency: Clinical, Biochemical and Genetic Features in 371 Patients". Molecular Genetics and Metabolism. 105 (1): 34–43. doi:10.1016/j.ymgme.2011.09.032. ISSN 1096-7192. PMC 3754811. PMID 22079328.
- Natarajan, Niranjana; Tully, Hannah M.; Chapman, Teresa (2016-08-01). "Prenatal presentation of pyruvate dehydrogenase complex deficiency". Pediatric Radiology. 46 (9): 1354–1357. doi:10.1007/s00247-016-3585-z. ISSN 1432-1998. PMC 6383724. PMID 27026023.
- Mitochondrion.
- "Pyruvate Dehydrogenase Complex Deficiency Workup: Laboratory Studies, Imaging Studies, Histologic Findings". emedicine.medscape.com. Retrieved 8 November 2016.
- Garry Brown, ed. (April 2012). "Pyruvate dehydrogenase deficiency". Orphanet. ORPHA765. Retrieved 8 November 2016.
- Sofou, Kalliopi; Dahlin, Maria; Hallböök, Tove; Lindefeldt, Marie; Viggedal, Gerd; Darin, Niklas (2017). "Ketogenic diet in pyruvate dehydrogenase complex deficiency: short- and long-term outcomes". Journal of Inherited Metabolic Disease. 40 (2): 237–245. doi:10.1007/s10545-016-0011-5. ISSN 1573-2665. PMC 5306430. PMID 28101805.
- "KetoCal family of ketogenic medical foods (ketogenic formulas)". www.myketocal.com. Retrieved 2020-05-06.
- "Metabolic Acidosis Treatment & Management: Approach Considerations, Type 1 Renal Tubular Acidosis, Type 2 Renal Tubular Acidosis". 2019-11-11. Cite journal requires
|journal=(help) - "Natural History and Advanced Genetic Study of Pyruvate Dehydrogenase Complex Deficiencies - Full Text View - ClinicalTrials.gov". clinicaltrials.gov. Retrieved 2020-05-06.
External links
| Classification | |
|---|---|
| External resources |