Reversible-deactivation radical polymerization
Reversible deactivation radical polymerizations are members of the class of reversible deactivation polymerizations which exhibit much of the character of living polymerizations, but cannot be categorized as such as they are not without chain transfer or chain termination reactions.[2][3] Several different names have been used in literature, which are:
- Living radical polymerization
- Living free radical polymerization
- Controlled/"living" radical polymerization
- Controlled radical polymerization
- Reversible deactivation radical polymerization
reversible-deactivation radical polymerization
See also reversible-deactivation polymerization RDP.
Though the term "living" radical polymerization was used in early days, it has been discouraged by IUPAC, because radical polymerization cannot be a truly living process due to unavoidable termination reactions between two radicals. The commonly used term controlled radical polymerization is permitted, but reversible-deactivated radical polymerization or controlled reversible-deactivation radical polymerization (RDRP) is recommended.
History and character
RDRP – sometimes misleadingly called 'free' radical polymerization – is one of the most widely used polymerization processes since it can be applied
- to a great variety of monomers
- it can be carried out in the presence of certain functional groups
- the technique is rather simple and easy to control
- the reaction conditions can vary from bulk over solution, emulsion, miniemulsion to suspension
- it is relatively inexpensive compared with competitive techniques
The steady-state concentration of the growing polymer chains is 10−7 M by order of magnitude, and the average life time of an individual polymer radical before termination is about 5–10 s. A drawback of the conventional radical polymerization is the limited control of chain architecture, molecular weight distribution, and composition. In the late 20th century it was observed that when certain components were added to systems polymerizing by a chain mechanism they are able to react reversibly with the (radical) chain carriers, putting them temporarily into a 'dormant' state.[4] [5] This had the effect of prolonging the lifetime of the growing polymer chains (see above) to values comparable with the duration of the experiment. At any instant most of the radicals are in the inactive (dormant) state, however, they are not irreversibly terminated (‘dead’). Only a small fraction of them are active (growing), yet with a fast rate of interconversion of active and dormant forms, faster than the growth rate, the same probability of growth is ensured for all chains, i.e., on average, all chains are growing at the same rate. Consequently, rather than a most probable distribution, the molecular masses (degrees of polymerization) assume a much narrower Poisson distribution, and a lower dispersity prevails.
IUPAC also recognizes the alternative name, ‘controlled reversible-deactivation radical polymerization’ as acceptable, "provided the controlled context is specified, which in this instance comprises molecular mass and molecular mass distribution." These types of radical polymerizations are not necessarily ‘living’ polymerizations, since chain termination reactions are not precluded".[1][2][3]
The adjective ‘controlled’ indicates that a certain kinetic feature of a polymerization or structural aspect of the polymer molecules formed is controlled (or both). The expression ‘controlled polymerization’ is sometimes used to describe a radical or ionic polymerization in which reversible-deactivation of the chain carriers is an essential component of the mechanism and interrupts the propagation that secures control of one or more kinetic features of the polymerization or one or more structural aspects of the macromolecules formed, or both. The expression ‘controlled radical polymerization’ is sometimes used to describe a radical polymerization that is conducted in the presence of agents that lead to e.g. atom-transfer radical polymerization (ATRP), nitroxide-(aminoxyl) mediated polymerization (NMP), or reversible-addition-fragmentation chain transfer (RAFT) polymerization. All these and further controlled polymerizations are included in the class of reversible-deactivation radical polymerizations. Whenever the adjective ‘controlled’ is used in this context the particular kinetic or the structural features that are controlled have to be specified.
Reversible-deactivation polymerization
There is a mode of polymerization referred to as reversible-deactivation polymerization which is distinct from living polymerization, despite some common features. Living polymerization requires a complete absence of termination reactions, whereas reversible-deactivation polymerization may contain a similar fraction of termination as conventional polymerization with the same concentration of active species.[1] Some important aspects of these are compared in the table:
| Property | Standard radical polymerization | Living polymerization | Reversible-deactivation polymerization |
|---|---|---|---|
| Concn. of initiating species | Falls off only slowly | Falls off very rapidly | Falls off very rapidly |
| Concn. of chain carriers (Number of growing chains) |
Instantaneous steady state (Bodenstein approximation applies) decreasing throughout reaction |
Constant throughout reaction | Constant throughout reaction |
| Lifetime of growing chains | ~ 10−3 s | Same as reaction duration | Same as reaction duration |
| Main form of termination | Radical combination or radical disproportionation |
Termination reactions are precluded | Termination reactions are not precluded |
| Molar mass distribution | Broad range (Ð >=1.5), Schulz-Zimm distribution |
Narrow range(Ð <1.5), Poisson distribution |
Narrow range(Ð <1.5), Poisson distribution |
| Dormant states | None | Rare | Predominant |
Common features
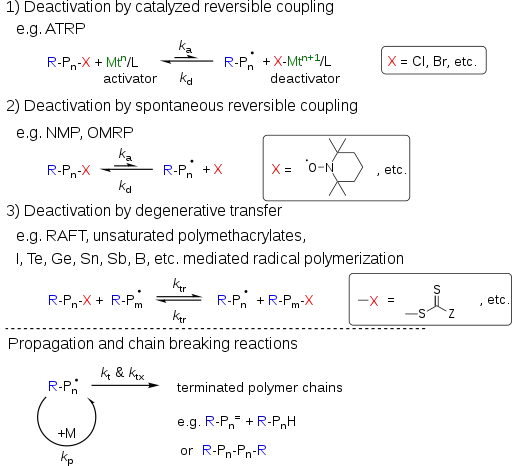
As the name suggests, the prerequisite of a successful RDRP is fast and reversible activation/deactivation of propagating chains. There are three types of RDRP; namely deactivation by catalyzed reversible coupling, deactivation by spontaneous reversible coupling and deactivation by degenerative transfer (DT). A mixture of different mechanisms is possible; e.g. a transition metal mediated RDRP could switch among ATRP, OMRP and DT mechanisms depending on the reaction conditions and reagents used.
In any RDRP processes, the radicals can propagate with the rate coefficient kp by addition of a few monomer units before the deactivation reaction occurs to regenerate the dormant species. Concurrently, two radicals may react with each other to form dead chains with the rate coefficient kt. The rates of propagation and termination between two radicals are not influenced by the mechanism of deactivation or the catalyst used in the system. Thus it is possible to estimate how fast a RDRP can be conducted with preserved chain end functionality?[6]
In addition, other chain breaking reactions such as irreversible chain transfer/termination reactions of the propagating radicals with solvent, monomer, polymer, catalyst, additives, etc. would introduce additional loss of chain end functionality (CEF).[7] The overall rate coefficient of chain breaking reactions besides the direct termination between two radicals is represented as ktx.
In all RDRP methods, the theoretical number average molecular weight of obtained polymers, Mn, can be defined by following equation:

where Mm is the molecular weight of monomer; [M]0 and [M]t are the monomer concentrations at time 0 and time t; [R-X]0 is the initial concentration of the initiator.
Besides the designed molecular weight, a well controlled RDRP should give polymers with narrow molecular distributions, which can be quantified by Mw/Mn values, and well preserved chain end functionalities.

A well controlled RDRP process requires: 1) the reversible deactivation process should be sufficiently fast; 2) the chain breaking reactions which cause the loss of chain end functionalities should be limited; 3) properly maintained radical concentration; 4) the initiator should have proper activity.
Examples
Atom transfer radical polymerization (ATRP)
The initiator of the polymerization is usually an organohalogenid and the dormant state is achieved in a metal complex of a transition metal (‘radical buffer’). This method is very versatile but requires unconventional initiator systems that are sometimes poorly compatible with the polymerization media.
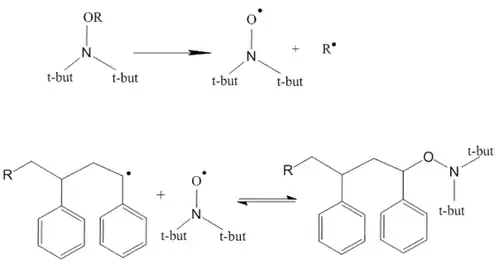
Nitroxide-mediated polymerization (NMP)
Given certain conditions a homolytic splitting of the C-O bond in alkoxylamines can occur and a stable 2-centre 3 electron N-O radical can be formed that is able to initiate a polymerization reaction. The preconditions for an alkoxylamine suitable to initiate a polymerization are bulky, sterically obstructive substituents on the secondary amine, and the substituent on the oxygen should be able to form a stable radical, e.g. benzyl.
Reversible addition-fragmentation chain transfer (RAFT)
RAFT is one of the most versatile and convenient techniques in this context. The most common RAFT-processes are carried out in the presence of thiocarbonylthio compounds that act as radical buffers. While in ATRP and NMP reversible deactivation of propagating radical-radical reactions takes place and the dormant structures are a halo-compound in ATRP and the alkoxyamine in NMP, both being a sink for radicals and source at the same time and described by the corresponding equilibria. RAFT on the contrary, is controlled by chain-transfer reactions that are in a deactivation-activation equilibrium. Since no radicals are generated or destroyed an external source of radicals is necessary for initiation and maintenance of the propagation reaction.
- Initiation step of a RAFT polymerization
- Reversible chain transfer
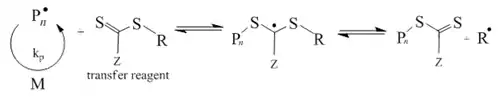
- Reinitiation step
- Chain equilibration step
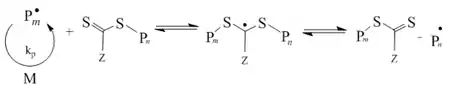
- Termination step



Catalytic chain transfer and cobalt mediated radical polymerization
Although not a strictly living form of polymerization catalytic chain transfer polymerization must be mentioned as it figures significantly in the development of later forms of living free radical polymerization. Discovered in the late 1970s in the USSR it was found that cobalt porphyrins were able to reduce the molecular weight during polymerization of methacrylates. Later investigations showed that the cobalt glyoxime complexes were as effective as the porphyrin catalysts and also less oxygen sensitive. Due to their lower oxygen sensitivity these catalysts have been investigated much more thoroughly than the porphyrin catalysts.
The major products of catalytic chain transfer polymerization are vinyl-terminated polymer chains. One of the major drawbacks of the process is that catalytic chain transfer polymerization does not produce macromonomers but instead produces addition fragmentation agents. When a growing polymer chain reacts with the addition fragmentation agent the radical end-group attacks the vinyl bond and forms a bond. However, the resulting product is so hindered that the species undergoes fragmentation, leading eventually to telechelic species.
These addition fragmentation chain transfer agents do form graft copolymers with styrenic and acrylate species however they do so by first forming block copolymers and then incorporating these block copolymers into the main polymer backbone.
While high yields of macromonomers are possible with methacrylate monomers, low yields are obtained when using catalytic chain transfer agents during the polymerization of acrylate and stryenic monomers. This has been seen to be due to the interaction of the radical centre with the catalyst during these polymerization reactions.
The reversible reaction of the cobalt macrocycle with the growing radical is known as cobalt carbon bonding and in some cases leads to living polymerization reactions.
Iniferter polymerization
An iniferter is a chemical compound that simultaneously acts as initiator, transfer agent, and terminator (hence the name ini-fer-ter) in controlled free radical iniferter polymerizations, the most common is the dithiocarbamate type.[8][9]
Iodine-transfer polymerization (ITP)
Iodine-transfer polymerization (ITP, also called ITRP), developed by Tatemoto and coworkers in the 1970s[10] gives relatively low polydispersities for fluoroolefin polymers. While it has received relatively little academic attention, this chemistry has served as the basis for several industrial patents and products and may be the most commercially successful form of living free radical polymerization.[11] It has primarily been used to incorporate iodine cure sites into fluoroelastomers.
The mechanism of ITP involves thermal decomposition of the radical initiator (typically persulfate), generating the initiating radical In•. This radical adds to the monomer M to form the species P1•, which can propagate to Pm•. By exchange of iodine from the transfer agent R-I to the propagating radical Pm• a new radical R• is formed and Pm• becomes dormant. This species can propagate with monomer M to Pn•. During the polymerization exchange between the different polymer chains and the transfer agent occurs, which is typical for a degenerative transfer process.
Typically, iodine transfer polymerization uses a mono- or diiodo-perfluoroalkane as the initial chain transfer agent. This fluoroalkane may be partially substituted with hydrogen or chlorine. The energy of the iodine-perfluoroalkane bond is low and, in contrast to iodo-hydrocarbon bonds, its polarization small.[12] Therefore, the iodine is easily abstracted in the presence of free radicals. Upon encountering an iodoperfluoroalkane, a growing poly(fluoroolefin) chain will abstract the iodine and terminate, leaving the now-created perfluoroalkyl radical to add further monomer. But the iodine-terminated poly(fluoroolefin) itself acts as a chain transfer agent. As in RAFT processes, as long as the rate of initiation is kept low, the net result is the formation of a monodisperse molecular weight distribution.
Use of conventional hydrocarbon monomers with iodoperfluoroalkane chain transfer agents has been described.[13] The resulting molecular weight distributions have not been narrow since the energetics of an iodine-hydrocarbon bond are considerably different from that of an iodine-fluorocarbon bond and abstraction of the iodine from the terminated polymer difficult. The use of hydrocarbon iodides has also been described, but again the resulting molecular weight distributions were not narrow.[14]
Preparation of block copolymers by iodine-transfer polymerization was also described by Tatemoto and coworkers in the 1970s.[15]
Although use of living free radical processes in emulsion polymerization has been characterized as difficult,[16] all examples of iodine-transfer polymerization have involved emulsion polymerization. Extremely high molecular weights have been claimed.[17]
Listed below are some other less described but to some extent increasingly important living radical polymerization techniques.
Selenium-centered radical-mediated polymerization
Diphenyl diselenide and several benzylic selenides have been explored by Kwon et al. as photoiniferters in polymerization of styrene and methyl methacrylate. Their mechanism of control over polymerization is proposed to be similar to the dithiuram disulfide iniferters. However, their low transfer constants allow them to be used for block copolymer synthesis but give limited control over the molecular weight distribution.[18]
Telluride-mediated polymerization (TERP)
Telluride-mediated polymerization or TERP first appeared to mainly operate under a reversible chain transfer mechanism by homolytic substitution under thermal initiation. However, in a kinetic study it was found that TERP predominantly proceeds by degenerative transfer rather than 'dissociation combination'.[19]
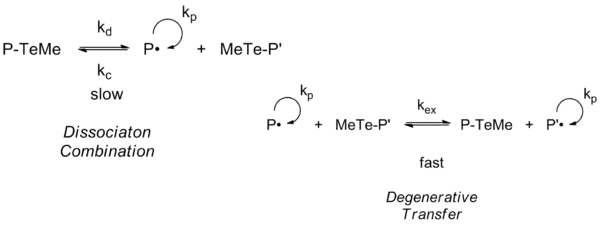
Alkyl tellurides of the structure Z-X-R, were Z=methyl and R= a good free radical leaving group, give the better control for a wide range of monomers, phenyl tellurides (Z=phenyl) giving poor control. Polymerization of methyl methacrylates are only controlled by ditellurides. The importance of X to chain transfer increases in the series O<S<Se<Te, makes alkyl tellurides effective in mediating control under thermally initiated conditions and the alkyl selenides and sulfides effective only under photoinitiated polymerization.
Stibine-mediated polymerization
More recently Yamago et al. reported stibine-mediated polymerization, using an organostibine transfer agent with the general structure Z(Z')-Sb-R (where Z= activating group and R= free radical leaving group). A wide range of monomers (styrenics, (meth)acrylics and vinylics) can be controlled, giving narrow molecular weight distributions and predictable molecular weights under thermally initiated conditions.[20][21] Yamago has also published a patent indicating that bismuth alkyls can also control radical polymerizations via a similar mechanism.
Copper mediated polymerization
More reversible-deactivation radical polymerizations are known to be catalysed by copper.
References
- Jenkins AD, Jones RG, Moad G (2009). "Terminology for reversible-deactivation radical polymerization previously called "controlled" radical or "living" radical polymerization (IUPAC Recommendations 2010)". Pure and Applied Chemistry. 82 (2): 483–491. doi:10.1351/PAC-REP-08-04-03. ISSN 1365-3075.
- Szwarz, M. (1956). "'Living' Polymers". Nature. 178 (1): 1168–1169. Bibcode:1956Natur.178.1168S. doi:10.1038/1781168a0.
- Szwarz, M. (2000). "Comments on "Living Polymerization: Rationale for Uniform Terminology" by Darling et al". J. Polym. Sci. A. 38 (10): 1710.
- US patent 4581429, D. H. Solomon, E. Rizzardo, P. Cacioli, "title"
- G. Moad; E. Rizzardo (1995). "Alkoxyamine-Initiated Living Radical Polymerization: Factors Affecting Alkoxyamine Homolysis Rates". Macromolecules. 28 (26): 8722–8728. Bibcode:1995MaMol..28.8722M. doi:10.1021/MA00130A003.
- Zhong, M; Matyjaszewski, K (2011). "How Fast Can a CRP Be Conducted with Preserved Chain End Functionality?". Macromolecules. 44 (8): 2668–2677. Bibcode:2011MaMol..44.2668Z. doi:10.1021/ma102834s.
- Wang, Y; Soerensen, N; Zhong, M; Schroeder, H; Buback, M; Matyjaszewski, K (2013). "Improving the "Livingness" of ATRP by Reducing Cu Catalyst Concentration". Macromolecules. 46 (3): 683–691. Bibcode:2013MaMol..46..683W. doi:10.1021/ma3024393.
- Otsu, Takayuki (1982). "Role of initiator-transfer agent-terminator (iniferter) in radical polymerizations: Polymer design by organic disulfides as iniferters". Die Makromolekulare Chemie, Rapid Communications. 3: 127–132. doi:10.1002/marc.1982.030030208.
- Otsu, Takayuki (1982). "A model for living radical polymerization". Die Makromolekulare Chemie, Rapid Communications. 3: 133–140. doi:10.1002/marc.1982.030030209.
- Tatemoto, Masayoshi; Suzuki, Takeshi; Tomoda, Masayasu; Furukawa, Yasuyoshi and Ueta, Yutaka (1981-01-06) "Cross linkable fluorine-containing polymer and its production" U.S. Patent 4,243,770.
- Ameduri, B; Boutevin, B (1999). "Use of telechelic fluorinated diiodides to obtain well-defined fluoropolymers". J. Fluorine Chem. 100: 97–116. doi:10.1016/s0022-1139(99)00220-1.
- Carlson, Dana P. (1991-08-06) "Base resistant fluoroelastomers with improved processibility and curability". U.S. Patent 5,037,921.
- Arcella, Vincenzo; Brinati, Giulio; Albano, Margherita and Tortelli, Vito (1996-12-17) "Fluoroelastomers comprising monomeric units deriving from a bis-olefin". U.S. Patent 5,585,449.
- Banus, J.; Emeleus, H. J.; Haszeldine, R. N. (1951). "12. The heterolytic fission of the carbon?iodine bond in trifluoroiodomethane". J. Chem. Soc.: 60. doi:10.1039/jr9510000060.
- Lansalot, M.; Farcet, C.; Charleux, B.; Vairon, J.-P. (1999). "Controlled Free-Radical Miniemulsion Polymerization of Styrene Using Degenerative Transfer". Macromolecules. 32 (22): 7354–7360. Bibcode:1999MaMol..32.7354L. doi:10.1021/ma990447w.
- Matyjaszewski, K.; Gaynor, S.; Wang, J.-S. (1995). "Controlled Radical Polymerizations: The Use of Alkyl Iodides in Degenerative Transfer". Macromolecules. 28 (6): 2093–2095. Bibcode:1995MaMol..28.2093M. doi:10.1021/ma00110a050.
- Ziegler, K (1936). "Die Bedeutung der alkalimetallorganischen Verbindungen für die Synthese". Angew. Chem. 49 (30): 499–502. doi:10.1002/ange.19360493003.
- Kwon, Tae Seok; Kumazawa, Sadanori; Yokoi, Tetsuya; Kondo, Shuji; Kunisada, Hideo; Yuki, Yasuo (1997). "Living Radical Polymerization of Styrene with Diphenyl Diselenide as a Photoiniferter. Synthesis of Polystyrene with Carbon-Carbon Double Bonds at Both Chain Ends". Journal of Macromolecular Science, Part A. 34 (9): 1553–1567. doi:10.1080/10601329708010026.
- Goto A, Kwak Y, Fukuda T, Yamago S, Iida K, Nakajima M, Yoshida J (2003). "Mechanism-based invention of high-speed living radical polymerization using organotellurium compounds and azo-initiators". J. Am. Chem. Soc. 125 (29): 8720–1. doi:10.1021/ja035464m. PMID 12862455.
- Yamago S, Ray B, Iida K, Yoshida J, Tada T, Yoshizawa K, Kwak Y, Goto A, Fukuda T (2004). "Highly versatile organostibine mediators for living radical polymerization". J. Am. Chem. Soc. 126 (43): 13908–9. doi:10.1021/ja044787v. PMID 15506736.
- Yamago S, Kayahara E, Kotani M, Ray B, Kwak Y, Goto A, Fukuda T (2007). "Highly controlled living radical polymerization through dual activation of organobismuthines". Angew. Chem. Int. Ed. Engl. 46 (8): 1304–6. doi:10.1002/anie.200604473. PMID 17205592.
