Structural bioinformatics
Structural bioinformatics is the branch of bioinformatics that is related to the analysis and prediction of the three-dimensional structure of biological macromolecules such as proteins, RNA, and DNA. It deals with generalizations about macromolecular 3D structures such as comparisons of overall folds and local motifs, principles of molecular folding, evolution, and binding interactions, and structure/function relationships, working both from experimentally solved structures and from computational models. The term structural has the same meaning as in structural biology, and structural bioinformatics can be seen as a part of computational structural biology. Structural bioinformatics main objectives are the creation of new methods to deal with biological macromolecules data to solve problems in biology and generate new knowledge.[1]
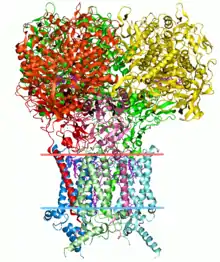
Introduction
Protein structure
The protein structure is directly related to its function. The presence of some chemical groups in specific locations allows proteins to act as enzymes, catalyzing several chemical reactions.[2] In general, protein structures are classified into four levels: primary (sequences), secondary (local conformation of the polypeptide chain), tertiary (three-dimensional structure of the protein fold), and quaternary (association of multiple polypeptide structures). Structural bioinformatics mainly addresses interactions among structures taking into consideration their space coordinates. Thus, the primary structure is better analyzed in traditional branches of bioinformatics. However, the sequence implies restrictions that allow the formation of conserved local conformations of the polypeptide chain, such as alpha-helix, beta-sheets, and loops (secondary structure[3]). Also, weak interactions (as hydrogen bonds) stabilize the protein fold. Interactions could be intrachain, i.e., when occurring between parts of the same protein monomer (tertiary structure), or interchain, i.e., when occurring between different structures (quaternary structure).
Structure visualization
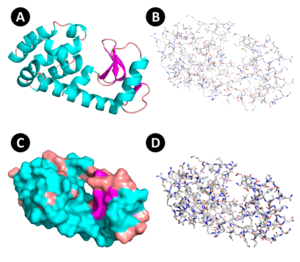
Protein structure visualization is an important issue for structural bioinformatics.[4] It allows users to observe static or dynamic representations of the molecules, also allowing the detection of interactions that could be used to infer about molecular mechanisms studied. The most common types of visualization are:
- Cartoon: this type of protein visualization highlights the secondary structure differences. In general, α-helix is represented as a type of screw, β-strands as arrows, and loops as lines.
- Lines: each amino acid residue is represented by thin lines, which allows a low cost for graphic rendering.
- Surface: in this visualization, the external shape of the molecule is shown.
- Sticks: each covalent bond between amino acid atoms is represented as a stick. This type of visualization is most used to visualize interactions between amino acids.
DNA structure
The classic DNA duplexes structure was initially described by Watson and Crick (and contributions of Rosalind Franklin). The DNA molecule is composed of three substances: a phosphate group, a pentose, and a nitrogen base (adenine, thymine, cytosine, or guanine). The DNA double helix structure is stabilized by hydrogen bonds formed between base pairs: adenine with thymine (A-T) and cytosine with guanine (C-G). Many structural bioinformatics studies have focused on understanding interactions between DNA and small molecules, which has been the target of several drug design studies.
Interactions
Interactions are contacts established between parts of molecules at different levels. They are responsible for stabilizing protein structures and perform a varied range of activities. In biochemistry, interactions are characterized by the proximity of atom groups or molecules regions that present an effect upon one another, such as electrostatic forces, hydrogen bonding, and hydrophobic effect. Proteins can perform several types of interactions, such as protein-protein interactions (PPI), protein-peptide interactions[5], protein-ligand interactions (PLI)[6], and protein-DNA interaction.
.png.webp)
Calculating contacts
Calculating contacts is an important task in structural bioinformatics, being important for the correct prediction of protein structure and folding, thermodynamic stability, protein-protein and protein-ligand interactions, docking and molecular dynamics analyses, and so on.[8]
Traditionally, computational methods have used threshold distance between atoms (also called cutoff) to detect possible interactions.[9] This detection is performed based on Euclidean distance and angles between atoms of determined types. However, most of the methods based on simple Euclidean distance cannot detect occluded contacts. Hence, cutoff free methods, such as Delaunay triangulation, have gained prominence in recent years. In addition, the combination of a set of criteria, for example, physicochemical properties, distance, geometry, and angles, have been used to improve the contact determination.[8]
| Type | Max distance criteria |
|---|---|
| Hydrogen bond | 3,9 Å |
| Hydrophobic interaction | 5 Å |
| Ionic interaction | 6 Å |
| Aromatic Stacking | 6 Å |
Protein Data Bank (PDB)
.png.webp)
The Protein Data Bank (PDB) is a database of 3D structure data for large biological molecules, such as proteins, DNA, and RNA. PDB is managed by an international organization called the Worldwide Protein Data Bank (wwPDB), which is composed of several local organizations, as. PDBe, PDBj, RCSB, and BMRB. They are responsible for keeping copies of PDB data available on the internet at no charge. The number of structure data available at PDB has increased each year, being obtained typically by X-ray crystallography, NMR spectroscopy, or cryo-electron microscopy.
Data format
The PDB format (.pdb) is the legacy textual file format used to store information of three-dimensional structures of macromolecules used by the Protein Data Bank. Due to restrictions in the format structure conception, the PDB format does not allow large structures containing more than 62 chains or 99999 atom records.[10]
The PDBx/mmCIF (macromolecular Crystallographic Information File) is a standard text file format for representing crystallographic information.[11] Since 2014, the PDB format was substituted as the standard PDB archive distribution by the PDBx/mmCIF file format (.cif). While PDB format contains a set of records identified by a keyword of up six characters, the PDBx/mmCIF format uses a structure based on key and value, where the key is a name that identifies some feature and the value is the variable information.[12]
Other structural databases
In addition to the Protein Data Bank (PDB), there are several databases of protein structures and other macromolecules. Examples include:
- MMDB: Experimentally determined three-dimensional structures of biomolecules derived from Protein Data Bank (PDB).[13]
- Nucleic acid Data Base (NDB): Experimentally determined information about nucleic acids (DNA, RNA).[14]
- Structural Classification of Proteins (SCOP): Comprehensive description of the structural and evolutionary relationships between structurally known proteins.[15]
- TOPOFIT-DB: Protein structural alignments based on the TOPOFIT method.[16]
- Electron Density Server (EDS): Electron-density maps and statistics about the fit of crystal structures and their maps.[17]
- CASP: Prediction Center Community-wide, worldwide experiment for protein structure prediction CASP.[18]
- PISCES server for creating non-redundant lists of proteins: Generates PDB list by sequence identity and structural quality criteria.[19]
- The Structural Biology Knowledgebase: Tools to aid in protein research design.[20]
- ProtCID: The Protein Common Interface Database Database of similar protein-protein interfaces in crystal structures of homologous proteins.[21]
Structures comparison
Structural alignment
Structural alignment is a method for comparison between 3D structures based on their shape and conformation.[22] It could be used to infer the evolutionary relationship among a set of proteins even with low sequence similarity. Structural alignment implies in superimpose a 3D structure under a second one, rotating and translating atoms in corresponding positions (in general, using the Cα atoms or even the backbone heavy atoms C, N, O, and Cα). Usually, the alignment quality is evaluated based on the root-mean-square deviation (RMSD) of atomic positions, i.e., the average distance between atoms after superimposition:

where δi is the distance between atom i and either a reference atom corresponding in the other structure or the mean coordinate of the N equivalent atoms. In general, the RMSD outcome is measured in Ångström (Å) unit, which is equivalent to 10−10 m. The nearer to zero the RMSD value, the more similar are the structures.
Graph-based structural signatures
Structural signatures, also called fingerprints, are macromolecule pattern representations that can be used to infer similarities and differences. Comparisons among a large set of proteins using RMSD still is a challenge due to the high computational cost of structural alignments. Structural signatures based on graph distance patterns among atom pairs have been used to determine protein identifying vectors and to detect non-trivial information.[23] Furthermore, algebra linear and machine learning can be used for clustering protein signatures, detecting protein-ligand interactions, predicting ΔΔG, and proposing mutations based on Euclidean distance.[24]
Structure prediction
The atomic structures of molecules can be obtained by several methods, such as X-ray crystallography (XRC), NMR spectroscopy, and 3D electron microscopy; however, these processes can present high costs and sometimes some structures can be hardly established, such as membrane proteins. Hence, it is necessary to computational approaches for determining 3D structures of macromolecules. The structure prediction methods are classified into comparative modeling and de novo modeling.
Comparative modeling
Comparative modeling, also known as homology modeling, corresponds to the methodology to construct three-dimensional structures from an amino acid sequence of a target protein and a template with known structure. The literature has described that evolutionarily related proteins tend to present a conserved three-dimensional structure.[25] In addition, sequences of distantly related proteins with identity lower than 20% can present different folds.[26]
De novo modeling
In structural bioinformatics, de novo modeling, also known as ab initio modeling, refers to approaches for obtaining three-dimensional structures from sequences without the necessity of a homologous known 3D structure. Despite the new algorithms and methods proposed in the last years, de novo protein structure prediction is still considered one of the remain outstanding issues in modern science.[27]
Structure validation
After structure modeling, an additional step of structure validation is necessary since many of both comparative and 'de novo' modeling algorithms and tools use heuristics to try assembly the 3D structure, which can generate many errors. Some validation strategies consist of calculating energy scores and comparing them with experimentally determined structures. For example, the DOPE score is an energy score used by the MODELLER tool for determining the best model.[28]
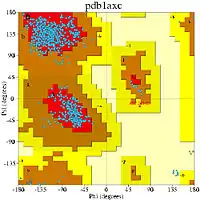
Another validation strategy is calculating φ and ψ backbone dihedral angles of all residues and construct a Ramachandran plot. The side-chain of amino acids and the nature of interactions in the backbone restrict these two angles, and thus, the visualization of allowed conformations could be performed based on the Ramachandran plot. A high quantity of amino acids allocated in no permissive positions of the chart is an indication of a low-quality modeling.
Prediction tools
A list with commonly used software tools for protein structure prediction, including comparative modeling, protein threading, de novo protein structure prediction, and secondary structure prediction is available in the list of protein structure prediction software.
Molecular docking
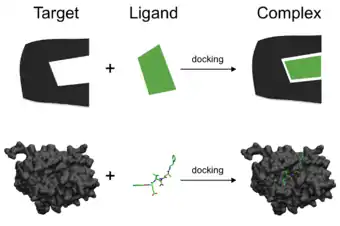
Molecular docking (also referred only as docking) is a method used to predict the orientation coordinates of a molecule (ligand) when linked to another one (receptor or target). Molecular docking aims to predict possible poses (binding modes) of the ligand when it interacts with specific regions, generally restricted by a box, in the receptor. Docking tools can use force fields to estimate a score for ranking best poses that favored better interactions.
In general, docking protocols are used to predict the interactions between small molecules and proteins. However, docking also can be used to detect associations and binding modes among proteins, peptides, DNA or RNA molecules, carbohydrates, and other macromolecules.
Virtual screening
Virtual screening (VS) is a computational approach used to fast screening of large compound libraries for drug discovery. Usually, virtual screening uses docking algorithms to rank small molecules with the highest affinity to a target receptor.
In recent times, several tools have been used to evaluate the use of virtual screening in the process of discovering new drugs. However, problems such as missing information, inaccurate understanding of drug-like molecules properties, weak scoring functions, or insufficient docking strategies hinder the docking process. Hence, the literature has described that it is still not considered a mature technology.[29][30]
Molecular dynamics
Molecular dynamics (MD) is a computational method for simulating interactions between molecules and their atoms during a given period of time.[32] This method allows the observation of the behavior of molecules and their interactions, considering the system as a whole. To calculate the behavior of the systems and, thus, determine the trajectories, an MD can use Newton's equation of motion, in addition to using molecular mechanics methods to estimate the forces that occur between particles (force fields).[33]
Applications
Informatics approaches used in structural bioinformatics are:
- Selection of Target - Potential targets are identified by comparing them with databases of known structures and sequence. The importance of a target can be decided on the basis of published literature. Target can also be selected on the basis of its protein domain. Protein domain are building blocks that can be rearranged to form new proteins. They can be studied in isolation initially.
- Tracking X-ray crystallography trials - X-Ray crystallography can be used to reveal three-dimensional structure of a protein. But, in order to use X-ray for studying protein crystals, pure proteins crystals must be formed, which can take a lot of trials. This leads to a need for tracking the conditions and results of trials. Furthermore, supervised machine learning algorithms can be used on the stored data to identify conditions that might increase the yield of pure crystals.
- Analysis of X-Ray crystallographic data - The diffraction pattern obtained as a result of bombarding X-rays on electrons is Fourier transform of electron density distribution. There is a need for algorithms that can deconvolve Fourier transform with partial information ( due to missing phase information, as the detectors can only measure amplitude of diffracted X-rays, and not the phase shifts ). Extrapolation technique such as Multiwavelength anomalous dispersion can be used to generate electron density map, which uses the location of selenium atoms as a reference to determine rest of the structure. Standard Ball-and-stick model is generated from the electron density map.
- Analysis of NMR spectroscopy data - Nuclear magnetic resonance spectroscopy experiments produce two (or higher) dimensional data, with each peak corresponding to a chemical group within the sample. Optimization methods are used to convert spectra into three dimensional structures.
- Correlating Structural information with functional information - Structural studies can be used as probe for structural-functional relationship.
Tools
| Software | Description |
|---|---|
| I-TASSER | Predicting three-dimensional structure model of protein molecules from amino acid sequences. |
| MOE | Molecular Operating Environment (MOE) is an extensive platform including structural modeling for proteins, protein families and antibodies[34] |
| SBL | The Structural Bioinformatics Library: end-user applications and advanced algorithms |
| BALLView | Molecular modeling and visualization[35] |
| STING | Visualization and analysis |
| PyMOL | Viewer and modeling[36] |
| VMD | Viewer, molecular dynamics[37] |
| KiNG | An open-source Java kinemage viewer |
| STRIDE | Determination of secondary structure from coordinates[38] |
| MolProbity | Structure-validation web server |
| PROCHECK | A structure-validation web service |
| CheShift | A protein structure-validation on-line application |
| 3D-mol.js | A molecular viewer for web applications developed using Javascript |
| PROPKA | Rapid prediction of protein pKa values based on empirical structure/function relationships |
| CARA | Computer Aided Resonance Assignment |
| Docking Server | A molecular docking web server |
| StarBiochem | A java protein viewer, features direct search of protein databank |
| SPADE | The structural proteomics application development environment |
| PocketSuite | A web portal for various web-servers for binding site-level analysis. PocketSuite is divided into:: PocketDepth (Binding site prediction)
PocketMatch (Binding site comparison), PocketAlign (Binding site alignment), and PocketAnnotate (Binding site annotation). |
| MSL | An open-source C++ molecular modeling software library for the implementation of structural analysis, prediction and design methods |
| PSSpred | Protein secondary structure prediction |
| Proteus | Webtool for suggesting mutation pairs |
| SDM | A server for predicting effects of mutations on protein stability |
See also
- MMDB
- Protein Data Bank
- Structural Classification of Proteins database
- STING
- Molecular modelling
- List of software for molecular mechanics modeling
- Molecular Conceptor
- Protein structure prediction
- CASP
References
- Gu J, Bourne PE (2011). Structural Bioinformatics (2nd ed.). Hoboken: John Wiley & Sons. ISBN 978-1-118-21056-7. OCLC 778339075.
- Gu J, Bourne PE (2009-03-16). Structural Bioinformatics. John Wiley & Sons. ISBN 978-0-470-18105-8.
- Kocincová L, Jarešová M, Byška J, Parulek J, Hauser H, Kozlíková B (February 2017). "Comparative visualization of protein secondary structures". BMC Bioinformatics. 18 (Suppl 2): 23. doi:10.1186/s12859-016-1449-z. PMC 5333176. PMID 28251875.
- Shi M, Gao J, Zhang MQ (July 2017). "Web3DMol: interactive protein structure visualization based on WebGL". Nucleic Acids Research. 45 (W1): W523–W527. doi:10.1093/nar/gkx383. PMC 5570197. PMID 28482028.
- Stanfield RL, Wilson IA (February 1995). "Protein-peptide interactions". Current Opinion in Structural Biology. 5 (1): 103–13. doi:10.1016/0959-440X(95)80015-S. PMID 7773739.
- Klebe G (2015). "Protein-ligand interactions as the basis for drug action.". In Scapin G, Patel D, Arnold E (eds.). Multifaceted Roles of Crystallography in Modern Drug Discovery. NATO Science for Peace and Security Series A: Chemistry and Biology. Dordrecht: Springer. pp. 83–92. doi:10.1007/978-3-642-17907-5_4. ISBN 978-3-642-17906-8.
- "Proteus | PROTein Engineering Supporter |". proteus.dcc.ufmg.br. Retrieved 2020-02-26.
- Martins PM, Mayrink VD, de Silveira S, da Silveira CH, de Lima LH, de Melo-Minardi RC (2018). "How to compute protein residue contacts more accurately?". Proceedings of the 33rd Annual ACM Symposium on Applied Computing - SAC '18. Pau, France: ACM Press: 60–67. doi:10.1145/3167132.3167136. ISBN 978-1-4503-5191-1. S2CID 49562347.
- da Silveira CH, Pires DE, Minardi RC, Ribeiro C, Veloso CJ, Lopes JC, et al. (February 2009). "Protein cutoff scanning: A comparative analysis of cutoff dependent and cutoff free methods for prospecting contacts in proteins" (PDF). Proteins. 74 (3): 727–43. doi:10.1002/prot.22187. PMID 18704933. S2CID 1208256.
- "PDBx/mmCIF General FAQ". mmcif.wwpdb.org. Retrieved 2020-02-26.
- wwPDB.org. "wwPDB: File Formats and the PDB". www.wwpdb.org. Retrieved 2020-02-26.
- "PDBx/mmCIF Dictionary Resources". mmcif.wwpdb.org. Retrieved 2020-02-26.
- "Macromolecular Structures Resource Group". www.ncbi.nlm.nih.gov. Retrieved 2020-04-13.
- "Nucleic Acid Database (NDB)". ndbserver.rutgers.edu. Retrieved 2020-04-13.
- "SCOP: Structural Classification of Proteins". 2007-09-11. Archived from the original on 2007-09-11. Retrieved 2020-04-13.
- Ilyin VA, Abyzov A, Leslin CM (July 2004). "Structural alignment of proteins by a novel TOPOFIT method, as a superimposition of common volumes at a topomax point". Protein Science. 13 (7): 1865–74. doi:10.1110/ps.04672604. PMC 2279929. PMID 15215530.
- "EDS - Uppsala Electron Density Server". eds.bmc.uu.se. Retrieved 2020-04-13.
- "Home - Prediction Center". www.predictioncenter.org. Retrieved 2020-04-13.
- ":: Dunbrack Lab". dunbrack.fccc.edu. Retrieved 2020-04-13.
- "Structural Biology KnowlegebaseSBKB - SBKB". sbkb.org. Retrieved 2020-04-13.
- "Protein Common Interface Database". dunbrack2.fccc.edu. Retrieved 2020-04-13.
- "Structural alignment (genomics)". ScienceDaily. Retrieved 2020-02-26.
- Pires DE, de Melo-Minardi RC, dos Santos MA, da Silveira CH, Santoro MM, Meira W (December 2011). "Cutoff Scanning Matrix (CSM): structural classification and function prediction by protein inter-residue distance patterns". BMC Genomics. 12 Suppl 4 (S4): S12. doi:10.1186/1471-2164-12-S4-S12. PMC 3287581. PMID 22369665.
- Mariano DC, Santos LH, Machado KD, Werhli AV, de Lima LH, de Melo-Minardi RC (January 2019). "A Computational Method to Propose Mutations in Enzymes Based on Structural Signature Variation (SSV)". International Journal of Molecular Sciences. 20 (2): 333. doi:10.3390/ijms20020333. PMC 6359350. PMID 30650542.
- Kaczanowski S, Zielenkiewicz P (March 2010). "Why similar protein sequences encode similar three-dimensional structures?" (PDF). Theoretical Chemistry Accounts. 125 (3–6): 643–650. doi:10.1007/s00214-009-0656-3. ISSN 1432-881X. S2CID 95593331.
- Chothia C, Lesk AM (April 1986). "The relation between the divergence of sequence and structure in proteins". The EMBO Journal. 5 (4): 823–6. doi:10.1002/j.1460-2075.1986.tb04288.x. PMC 1166865. PMID 3709526.
- "So much more to know". Science. 309 (5731): 78–102. July 2005. doi:10.1126/science.309.5731.78b. PMID 15994524.
- Webb B, Sali A (September 2014). "Comparative Protein Structure Modeling Using MODELLER". Current Protocols in Bioinformatics. 47 (1): 5.6.1–32. doi:10.1002/0471250953.bi0506s47. PMC 4186674. PMID 25199792.
- Dhasmana A, Raza S, Jahan R, Lohani M, Arif JM (2019-01-01). "Chapter 19 - High-Throughput Virtual Screening (HTVS) of Natural Compounds and Exploration of Their Biomolecular Mechanisms: An In Silico Approach". In Ahmad Khan MS, Ahmad I, Chattopadhyay D (eds.). New Look to Phytomedicine. Academic Press. pp. 523–548. doi:10.1016/b978-0-12-814619-4.00020-3. ISBN 978-0-12-814619-4.
- Wermuth CG, Villoutreix B, Grisoni S, Olivier A, Rocher JP (January 2015). "Strategies in the search for new lead compounds or original working hypotheses.". In Wermuth CG, Aldous D, Raboisson P, Rognan D (eds.). The practice of Medicinal Chemistry. Academic Press. pp. 73–99. doi:10.1016/B978-0-12-417205-0.00004-3. ISBN 978-0-12-417205-0.
- Costa LS, Mariano DC, Rocha RE, Kraml J, Silveira CH, Liedl KR, et al. (September 2019). "Molecular Dynamics Gives New Insights into the Glucose Tolerance and Inhibition Mechanisms on β-Glucosidases". Molecules. 24 (18): 3215. doi:10.3390/molecules24183215. PMC 6766793. PMID 31487855.
- Alder BJ, Wainwright TE (August 1959). "Studies in Molecular Dynamics. I. General Method". The Journal of Chemical Physics. 31 (2): 459–466. Bibcode:1959JChPh..31..459A. doi:10.1063/1.1730376. ISSN 0021-9606.
- Yousif, Ragheed Hussam (2020). "Exploring the Molecular Interactions between Neoculin and the Human Sweet Taste Receptors through Computational Approaches" (PDF). Sains Malaysiana. 49 (3): 517–525. doi:10.17576/jsm-2020-4903-06.
- MOE
- BALLView
- PyMOL
- VMD
- STRIDE
Further reading
- Bourne PE, Gu J (2009). Structural Bioinformatics (2nd ed.). New York: John Wiley & Sons. ISBN 978-0-470-18105-8.
- Bourne PE, Weissig H (2003). Structural Bioinformatics. Wiley. ISBN 0-471-20199-5.
- Leach A (2001). Molecular Modelling: Principles and Applications (2nd ed.). Prentice Hall. ISBN 978-0-582-38210-7.
- Peitsch MC, Schwede T (2008). Computational Structural Biology: Methods and Applications. World Scientific. ISBN 978-9812778772.
- Leontis NB, Westhof E (April 2001). "Geometric nomenclature and classification of RNA base pairs". RNA. 7 (4): 499–512. doi:10.1017/S1355838201002515. PMC 1370104. PMID 11345429.
- Richardson JS (1981). "The anatomy and taxonomy of protein structure". Advances in Protein Chemistry Volume 34. Advances in Protein Chemistry. 34. pp. 167–339. doi:10.1016/S0065-3233(08)60520-3. ISBN 978-0-12-034234-1. PMID 7020376.
- Ramachandran GN, Sasisekharan V (1968). "Conformation of polypeptides and proteins". Advances in Protein Chemistry Volume 23. Advances in Protein Chemistry. 23. pp. 283–438. doi:10.1016/S0065-3233(08)60402-7. ISBN 978-0-12-034223-5. PMID 4882249.
- Ramachandran GN, Ramakrishnan C, Sasisekharan V (July 1963). "Stereochemistry of polypeptide chain configurations". Journal of Molecular Biology. 7: 95–9. doi:10.1016/S0022-2836(63)80023-6. PMID 13990617.
| Genomics | |
|---|---|
| Bioinformatics | |
| Structural biology | |
| Research tools | |
| Organizations | |
| |
