CLCN5
The CLCN5 gene encodes the chloride channel Cl-/H+ exchanger ClC-5. ClC-5 is mainly expressed in the kidney, in particular in proximal tubules where it participates to the uptake of albumin and low-molecular-weight proteins, which is one of the principal physiological role of proximal tubular cells. Mutations in the CLCN5 gene cause an X-linked recessive nephropathy named Dent disease (Dent disease 1 MIM#300009) characterized by excessive urinary loss of low-molecular-weight proteins and of calcium (hypercalciuria), nephrocalcinosis (presence of calcium phosphate aggregates in the tubular lumen and/or interstitium) and nephrolithiasis (kidney stones).

The CLCN5 gene
Structure
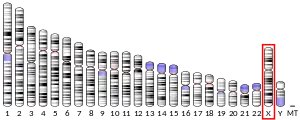
The human CLCN5 gene (MIM#300008, reference sequence NG_007159.2) is localized in the pericentromeric region on chromosome Xp11.23. It extends over about 170 Kb of genomic DNA, has a coding region of 2,238 bp and consists of 17 exons including 11 coding exons (from 2 to 12).[5][6][7][8] The CLCN5 gene has 8 paralogues (CLCN1, CLCN2, CLCN3, CLCN4, CLCN6, CLCN7, CLCNKA, CLCNKB) and 201 orthologues among jawed vertebrates (Gnathostomata).
Five different CLCN5 gene transcripts have been discovered, two of which (transcript variants 3 [NM_000084.5] and 4 [NM_001282163.1]) encode for the canonical 746 amino acid protein, two (transcript variants 1 [NM_001127899.3] and 2 [NM_001127898.3]) for the NH2-terminal extended 816 amino acid protein[9] and one does not encode for any protein (Transcript variant 5, [NM_001272102.2]). The 5’ untranslated region (5’UTR) of CLCN5 is complex and not entirely clarified. Two strong and one weak promoters were predicted to be present in the CLCN5 gene.[10][11] Several different 5’ alternatively used exons have been recognized in the human kidney.[9][10][11][12] The three promoters drive with varying degree of efficiency 11 different mRNAs, with transcription initiating from at least three different start sites.[10]
The chloride channel H+/Cl− exchanger ClC-5
Like all ClC channels, ClC-5 needs to dimerize to create the pore through which the ions pass.[13][14][15] ClC-5 can form both homo- and hetero-dimers due to its marked sequence homology with ClC-3 and ClC-4.[16][17][18]
The canonical 746-amino acid ClC-5 protein has 18 membrane spanning α-helices (named A to R), an intracellular N- terminal domain and a cytoplasmic C-terminus containing two cystathionine beta-synthase (CBS) domains which are known to be involved in the regulation of ClC-5 activity.[13][19][20][21] Helices B, H, I, O, P, and Q are the six major helices involved in the formation of dimer’s interface and are crucial for proper pore configuration.[13][14] The Cl− selectivity filter is principally driven by helices D, F, N, and R, which are conveyed together near the channel center.[13][14][22][23] Two important amino acids for the proper ClC-5 function are the glutamic acids at position 211 and 268 called respectively “gating glutamate” and “proton glutamate”.[24][25][26][27] The gating glutamate is necessary for both H+ transport and ClC-5 voltage dependence.[8][28][29] The proton glutamate is crucial to the H+ transport acting as an H+ transfer site.[24][30][31]
Localization and function
ClC-5 belongs to the family of voltage gated chloride channel that are regulators of membrane excitability, transepithelial transport and cell volume in different tissues. Based on sequence homology, the nine mammalian ClC proteins can be grouped into three classes, of which the first (ClC-1, ClC-2, ClC-Ka and ClC-Kb) is expressed primarily in plasma membranes, whereas the other two (ClC-3, ClC-4, and ClC-5 and ClC-6 and ClC-7) are expressed primarily in organellar membranes.[32]
ClC-5 is expressed in minor to moderate level in brain, muscle, intestine but highly in the kidney, primarily in proximal tubular cells of S3 segment, in alfa intercalated cells of cortical collecting duct of and in cortical and medullary thick ascending limb of Henle’s loop.[33][34][35][36][37][38]
Proximal tubular cells (PTCs) are the main site of ClC-5 expression. By means of the receptor-mediated endocytosis process, they uptake albumin and low-molecular-weight proteins freely passed through the glomerular filter. ClC-5 is located in early endosomes of PTCs where it co-localizes with the electrogenic vacuolar H+‐ATPase (V‐ATPase).[34][38] ClC-5 in this compartment contributes to the maintenance of intra-endosomal acidic pH. Environment acidification is necessary for the dissociation of ligand from its receptor. The receptor is then recycled to the apical membrane, while ligand is transported to the late endosome and lysosome where it is degraded. ClC-5 supports efficient acidification of endosomes either by providing a Cl− conductance to counterbalance the accumulation of positively charged H+ pumped in by V-ATPase or by directly acidifying endosome in parallel with V-ATPase.[39]
Experimental evidence indicates that endosomal Cl− concentration, which is raised by ClC-5 in exchange for protons accumulated by the V-ATPase, may play a role in endocytosis independently from endosomal acidification, thus pointing to another possible mechanism by which ClC-5 dysfunction may impair endocytosis.[40]
ClC-5 is located also at the cell surface of PTCs where probably it plays a role in the formation/function of the endocytic complex that also involves megalin and cubilin/amnionless receptors, the sodium-hydrogen antiporter 3 (NHE3), and the V-ATPase.[41][42] It was demonstrated at the C-terminus of ClC-5 binds the actin-depolymerizing protein cofilin. When the nascent endosome forms, the recruitment of cofilin by ClC-5 is a prerequisite for the localized dissolution of the actin cytoskeleton, thus permitting the endosome to pass into the cytoplasm. It is conceivable that at the cell surface, the large intracellular C-terminus of ClC-5 has a crucial function in mediating the assembly, stabilization and disassembly of the endocytic complex via protein–protein interactions. Therefore, ClC-5 may accomplish two roles in the receptor-mediated endocytosis: i) vesicular acidification and receptor recycling; ii) participation to the non-selective megalin–cubilin-amnionless low-molecular-weight protein uptake at the apical membrane.[41]
Clinical significance
Dent disease is mainly caused by loss-of-function mutations in the CLCN5 gene (Dent disease 1; MIM#300009).[14][43] Dent disease 1 shows a marked allelic heterogeneity. To date, 265 different CLCN5 pathogenic variants have been described.[14] A small number of pathogenic variants were found in more than one family.[44] The 48% are truncating mutations (nonsense, frameshift or complex), 37% non-truncating (missense or in-frame insertions/deletions), 10% splice site mutations, and 5% other type (large deletions, Alu insertions or 5’UTR mutations). Functional investigations in Xenopus Levis oocytes and mammalian cells[39][43][45][46][47][40] enabled these CLCN5 mutations to be classified according to their functional consequences.[8][44][48][49][50] The most common mutations lead to a defective protein folding and processing, resulting in endoplasmic reticulum retention of the mutant protein for further degradation by the proteasome.
Animal models
Two independent ClC-5 knock-out mice, the so called Jentsch[51][52] and Guggino models,[53][54][55][56] provided critical insights into the mechanisms of proximal tubular dysfunction in Dent disease 1. These two murine models recapitulated the major features of Dent disease (low-molecular-weight proteinuria, hypercalciuria and nephrocalcinosis/nephrolithiasis) and demonstrated that ClC-5 inactivation is associated with severe impairment of both fluid phase and receptor-mediated endocytosis, as well as trafficking defects leading to the loss of megalin and cubilin at the brush border of proximal tubules. However, targeted disruption of ClC-5 in the Jentsch model did not lead to hypercalciuria, kidney stones or nephrocalcinosis, while the Guggino model did.[53] The Jentsch murine model produced slightly more acidic urines. Urinary phosphate excretion was increased in both models by about 50%. Hyperphosphaturia in the Jentsch model was associated with decreased apical expression of the sodium/phosphate cotransporter NaPi2a that is the predominant phosphate transporter in the proximal tubule. However, NaPi2a expression is ClC-5-independent since apical NaPi2a was normally expressed in any proximal tubules of chimeric female mice, while it was decreased in all male proximal tubular knock-out cells. Serum parathormone (PTH) is normal in knock-out mice while urinary PTH is increased of about 1.7 fold. Megalin usually mediates the endocytosis and degradation of PTH in proximal tubular cells. In knock-out mice, the downregulation of megalin leads to PTH defective endocytosis and progressively increases luminal PTH levels that enhance the internalization of NaPi2a.[51]
DNA testing and genetic counselling
A clinical diagnosis of Dent disease can be confirmed through molecular genetic testing that can detect mutations in specific genes known to cause Dent disease. However, about 20-25% of Dent disease patients remain genetically unresolved.[44]
Genetic testing is useful to determine the status of healthy carrier in the mother of an affected male. In fact, being Dent disease an X-linked recessive disorder, males are more frequently affected than females, and females may be heterozygous healthy carrier. Due to skewed X-inactivation, female carriers may present some mild symptoms of Dent disease such as low-molecular-weight proteinuria or hypercalciuria. Carriers will transmit the disease to half of their sons whereas half of their daughters will be carriers. Affected males do not transmit the disease to their sons since they pass Y chromosome to males, but all their daughters will inherited mutated X chromosome. Preimplant and prenatal genetic testing is not advised for Dent disease 1 since the prognosis for the majority of the patients is good and a clear correlation between genotype and phenotype is lacking.[57]
See also
References
- GRCh38: Ensembl release 89: ENSG00000171365 - Ensembl, May 2017
- GRCm38: Ensembl release 89: ENSMUSG00000004317 - Ensembl, May 2017
- "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- Scheinman SJ, Pook MA, Wooding C, Pang JT, Frymoyer PA, Thakker RV (June 1993). "Mapping the gene causing X-linked recessive nephrolithiasis to Xp11.22 by linkage studies". The Journal of Clinical Investigation. 91 (6): 2351–7. doi:10.1172/JCI116467. PMC 443292. PMID 8099916.
- Fisher SE, Black GC, Lloyd SE, Hatchwell E, Wrong O, Thakker RV, Craig IW (November 1994). "Isolation and partial characterization of a chloride channel gene which is expressed in kidney and is a candidate for Dent's disease (an X-linked hereditary nephrolithiasis)". Human Molecular Genetics. 3 (11): 2053–9. PMID 7874126.
- Fisher SE, van Bakel I, Lloyd SE, Pearce SH, Thakker RV, Craig IW (October 1995). "Cloning and characterization of CLCN5, the human kidney chloride channel gene implicated in Dent disease (an X-linked hereditary nephrolithiasis)". Genomics. 29 (3): 598–606. doi:10.1006/geno.1995.9960. hdl:11858/00-001M-0000-0012-CC06-6. PMID 8575751.
- Scheel O, Zdebik AA, Lourdel S, Jentsch TJ (July 2005). "Voltage-dependent electrogenic chloride/proton exchange by endosomal CLC proteins". Nature. 436 (7049): 424–7. doi:10.1038/nature03860. PMID 16034422.
- Ludwig M, Waldegger S, Nuutinen M, Bökenkamp A, Reissinger A, Steckelbroeck S, Utsch B (2003). "Four additional CLCN5 exons encode a widely expressed novel long CLC-5 isoform but fail to explain Dent's phenotype in patients without mutations in the short variant". Kidney & Blood Pressure Research. 26 (3): 176–84. doi:10.1159/000071883. PMID 12886045.
- Hayama, Atsushi; Uchida, Shinichi; Sasaki, Sei; Marumo, Fumiaki (2000). "Isolation and characterization of the human CLC-5 chloride channel gene promoter". Gene. 261 (2): 355–364. doi:10.1016/S0378-1119(00)00493-5. PMID 11167024.
- Tosetto E, Casarin A, Salviati L, Familiari A, Lieske JC, Anglani F (July 2014). "Complexity of the 5'UTR region of the CLCN5 gene: eleven 5'UTR ends are differentially expressed in the human kidney". BMC Medical Genomics. 7 (1): 41. doi:10.1186/1755-8794-7-41. PMC 4105828. PMID 25001568.
- Forino M, Graziotto R, Tosetto E, Gambaro G, D'Angelo A, Anglani F (2004). "Identification of a novel splice site mutation of CLCN5 gene and characterization of a new alternative 5' UTR end of ClC-5 mRNA in human renal tissue and leukocytes". Journal of Human Genetics. 49 (1): 53–60. doi:10.1007/s10038-003-0108-1. PMID 14673707.
- Wu F, Roche P, Christie PT, Loh NY, Reed AA, Esnouf RM, Thakker RV (April 2003). "Modeling study of human renal chloride channel (hCLC-5) mutations suggests a structural-functional relationship". Kidney International. 63 (4): 1426–32. doi:10.1046/j.1523-1755.2003.00859.x. PMID 12631358.
- Mansour-Hendili L, Blanchard A, Le Pottier N, Roncelin I, Lourdel S, Treard C, et al. (August 2015). "Mutation Update of the CLCN5 Gene Responsible for Dent Disease 1" (PDF). Human Mutation. 36 (8): 743–52. doi:10.1002/humu.22804. PMID 25907713.
- Lourdel S, Grand T, Burgos J, González W, Sepúlveda FV, Teulon J (February 2012). "ClC-5 mutations associated with Dent's disease: a major role of the dimer interface" (PDF). Pflugers Archiv. 463 (2): 247–56. doi:10.1007/s00424-011-1052-0. PMID 22083641.
- Jentsch TJ, Günther W, Pusch M, Schwappach B (January 1995). "Properties of voltage-gated chloride channels of the ClC gene family". The Journal of Physiology. 482 (suppl): 19S–25S. doi:10.1113/jphysiol.1995.sp020560. PMC 1334233. PMID 7730971.
- Okkenhaug H, Weylandt KH, Carmena D, Wells DJ, Higgins CF, Sardini A (November 2006). "The human ClC-4 protein, a member of the CLC chloride channel/transporter family, is localized to the endoplasmic reticulum by its N-terminus". FASEB Journal. 20 (13): 2390–2. doi:10.1096/fj.05-5588fje. PMID 17023393.
- Mohammad-Panah R, Harrison R, Dhani S, Ackerley C, Huan LJ, Wang Y, Bear CE (August 2003). "The chloride channel ClC-4 contributes to endosomal acidification and trafficking". The Journal of Biological Chemistry. 278 (31): 29267–77. doi:10.1074/jbc.M304357200. PMID 12746443.
- Meyer S, Savaresi S, Forster IC, Dutzler R (January 2007). "Nucleotide recognition by the cytoplasmic domain of the human chloride transporter ClC-5". Nature Structural & Molecular Biology. 14 (1): 60–7. doi:10.1038/nsmb1188. PMID 17195847.
- Wellhauser L, Luna-Chavez C, D'Antonio C, Tainer J, Bear CE (February 2011). "ATP induces conformational changes in the carboxyl-terminal region of ClC-5". The Journal of Biological Chemistry. 286 (8): 6733–41. doi:10.1074/jbc.M110.175877. PMC 3057859. PMID 21173145.
- Zifarelli G, Pusch M (October 2009). "Intracellular regulation of human ClC-5 by adenine nucleotides". EMBO Reports. 10 (10): 1111–6. doi:10.1038/embor.2009.159. PMC 2759731. PMID 19713962.
- Grand T, Mordasini D, L'Hoste S, Pennaforte T, Genete M, Biyeyeme MJ, et al. (November 2009). "Novel CLCN5 mutations in patients with Dent's disease result in altered ion currents or impaired exchanger processing" (PDF). Kidney International. 76 (9): 999–1005. doi:10.1038/ki.2009.305. PMID 19657328.
- Pusch M, Ludewig U, Jentsch TJ (January 1997). "Temperature dependence of fast and slow gating relaxations of ClC-0 chloride channels". The Journal of General Physiology. 109 (1): 105–16. doi:10.1085/jgp.109.1.105. PMC 2217054. PMID 8997669.
- Zdebik AA, Zifarelli G, Bergsdorf EY, Soliani P, Scheel O, Jentsch TJ, Pusch M (February 2008). "Determinants of anion-proton coupling in mammalian endosomal CLC proteins". The Journal of Biological Chemistry. 283 (7): 4219–27. doi:10.1074/jbc.M708368200. PMID 18063579.
- Grieschat M, Alekov AK (March 2012). "Glutamate 268 regulates transport probability of the anion/proton exchanger ClC-5". The Journal of Biological Chemistry. 287 (11): 8101–9. doi:10.1074/jbc.M111.298265. PMC 3318731. PMID 22267722.
- Dutzler R, Campbell EB, MacKinnon R (April 2003). "Gating the selectivity filter in ClC chloride channels". Science. 300 (5616): 108–12. doi:10.1126/science.1082708. PMID 12649487.
- Yin J, Kuang Z, Mahankali U, Beck TL (November 2004). "Ion transit pathways and gating in ClC chloride channels". Proteins. 57 (2): 414–21. arXiv:physics/0401022. doi:10.1002/prot.20208. PMID 15340928.
- Friedrich T, Breiderhoff T, Jentsch TJ (January 1999). "Mutational analysis demonstrates that ClC-4 and ClC-5 directly mediate plasma membrane currents". The Journal of Biological Chemistry. 274 (2): 896–902. doi:10.1074/jbc.274.2.896. PMID 9873029.
- Picollo A, Pusch M (July 2005). "Chloride/proton antiporter activity of mammalian CLC proteins ClC-4 and ClC-5". Nature. 436 (7049): 420–3. doi:10.1038/nature03720. PMID 16034421.
- Accardi A, Walden M, Nguitragool W, Jayaram H, Williams C, Miller C (December 2005). "Separate ion pathways in a Cl-/H+ exchanger". The Journal of General Physiology. 126 (6): 563–70. doi:10.1085/jgp.200509417. PMC 2266597. PMID 16316975.
- Neagoe I, Stauber T, Fidzinski P, Bergsdorf EY, Jentsch TJ (July 2010). "The late endosomal ClC-6 mediates proton/chloride countertransport in heterologous plasma membrane expression". The Journal of Biological Chemistry. 285 (28): 21689–97. doi:10.1074/jbc.M110.125971. PMC 2898453. PMID 20466723.
- Stölting G, Fischer M, Fahlke C (2014-10-07). "CLC channel function and dysfunction in health and disease". Frontiers in Physiology. 5: 378. doi:10.3389/fphys.2014.00378. PMC 4188032. PMID 25339907.
- Obermüller N, Gretz N, Kriz W, Reilly RF, Witzgall R (February 1998). "The swelling-activated chloride channel ClC-2, the chloride channel ClC-3, and ClC-5, a chloride channel mutated in kidney stone disease, are expressed in distinct subpopulations of renal epithelial cells". The Journal of Clinical Investigation. 101 (3): 635–42. doi:10.1172/JCI1496. PMC 508607. PMID 9449697.
- Günther W, Lüchow A, Cluzeaud F, Vandewalle A, Jentsch TJ (July 1998). "ClC-5, the chloride channel mutated in Dent's disease, colocalizes with the proton pump in endocytotically active kidney cells". Proceedings of the National Academy of Sciences of the United States of America. 95 (14): 8075–80. doi:10.1073/pnas.95.14.8075. PMC 20931. PMID 9653142.
- Luyckx VA, Goda FO, Mount DB, Nishio T, Hall A, Hebert SC, et al. (November 1998). "Intrarenal and subcellular localization of rat CLC5". The American Journal of Physiology. 275 (5): F761-9. doi:10.1152/ajprenal.1998.275.5.F761. PMID 9815133.
- Lamb FS, Clayton GH, Liu BX, Smith RL, Barna TJ, Schutte BC (March 1999). "Expression of CLCN voltage-gated chloride channel genes in human blood vessels". Journal of Molecular and Cellular Cardiology. 31 (3): 657–66. doi:10.1006/jmcc.1998.0901. PMID 10198195.
- von Weikersthal SF, Barrand MA, Hladky SB (April 1999). "Functional and molecular characterization of a volume-sensitive chloride current in rat brain endothelial cells". The Journal of Physiology. 516 ( Pt 1): 75–84. doi:10.1111/j.1469-7793.1999.075aa.x. PMC 2269222. PMID 10066924.
- Devuyst O, Christie PT, Courtoy PJ, Beauwens R, Thakker RV (February 1999). "Intra-renal and subcellular distribution of the human chloride channel, CLC-5, reveals a pathophysiological basis for Dent's disease". Human Molecular Genetics. 8 (2): 247–57. doi:10.1093/hmg/8.2.247. PMID 9931332.
- Smith AJ, Lippiat JD (June 2010). "Direct endosomal acidification by the outwardly rectifying CLC-5 Cl(-)/H(+) exchanger". The Journal of Physiology. 588 (Pt 12): 2033–45. doi:10.1113/jphysiol.2010.188540. PMC 2911210. PMID 20421284.
- Novarino G, Weinert S, Rickheit G, Jentsch TJ (June 2010). "Endosomal chloride-proton exchange rather than chloride conductance is crucial for renal endocytosis". Science. 328 (5984): 1398–401. doi:10.1126/science.1188070. PMID 20430975.
- Hryciw DH, Wang Y, Devuyst O, Pollock CA, Poronnik P, Guggino WB (October 2003). "Cofilin interacts with ClC-5 and regulates albumin uptake in proximal tubule cell lines". The Journal of Biological Chemistry. 278 (41): 40169–76. doi:10.1074/jbc.M307890200. PMID 12904289.
- Wang Y, Cai H, Cebotaru L, Hryciw DH, Weinman EJ, Donowitz M, et al. (October 2005). "ClC-5: role in endocytosis in the proximal tubule" (PDF). American Journal of Physiology. Renal Physiology. 289 (4): F850-62. doi:10.1152/ajprenal.00011.2005. PMID 15942052.
- Lloyd SE, Pearce SH, Fisher SE, Steinmeyer K, Schwappach B, Scheinman SJ, et al. (February 1996). "A common molecular basis for three inherited kidney stone diseases". Nature. 379 (6564): 445–9. doi:10.1038/379445a0. PMID 8559248.
- Lieske, John C.; Milliner, Dawn S.; Beara-Lasic, Lada; Harris, Peter; Cogal, Andrea; Abrash, Elizabeth (1993), Adam, Margaret P.; Ardinger, Holly H.; Pagon, Roberta A.; Wallace, Stephanie E. (eds.), "Dent Disease", GeneReviews®, University of Washington, Seattle, PMID 22876375, retrieved 2020-02-28
- Lloyd SE, Gunther W, Pearce SH, Thomson A, Bianchi ML, Bosio M, et al. (August 1997). "Characterisation of renal chloride channel, CLCN5, mutations in hypercalciuric nephrolithiasis (kidney stones) disorders". Human Molecular Genetics. 6 (8): 1233–9. doi:10.1093/hmg/6.8.1233. PMID 9259268.
- Grand T, L'Hoste S, Mordasini D, Defontaine N, Keck M, Pennaforte T, et al. (April 2011). "Heterogeneity in the processing of CLCN5 mutants related to Dent disease" (PDF). Human Mutation. 32 (4): 476–83. doi:10.1002/humu.21467. PMID 21305656.
- Gorvin CM, Wilmer MJ, Piret SE, Harding B, van den Heuvel LP, Wrong O, et al. (April 2013). "Receptor-mediated endocytosis and endosomal acidification is impaired in proximal tubule epithelial cells of Dent disease patients". Proceedings of the National Academy of Sciences of the United States of America. 110 (17): 7014–9. doi:10.1073/pnas.1302063110. PMC 3637698. PMID 23572577.
- D'Antonio C, Molinski S, Ahmadi S, Huan LJ, Wellhauser L, Bear CE (June 2013). "Conformational defects underlie proteasomal degradation of Dent's disease-causing mutants of ClC-5". The Biochemical Journal. 452 (3): 391–400. doi:10.1042/BJ20121848. PMID 23566014.
- Smith AJ, Reed AA, Loh NY, Thakker RV, Lippiat JD (February 2009). "Characterization of Dent's disease mutations of CLC-5 reveals a correlation between functional and cell biological consequences and protein structure". American Journal of Physiology. Renal Physiology. 296 (2): F390-7. doi:10.1152/ajprenal.90526.2008. PMC 2643861. PMID 19019917.
- Ludwig M, Doroszewicz J, Seyberth HW, Bökenkamp A, Balluch B, Nuutinen M, et al. (July 2005). "Functional evaluation of Dent's disease-causing mutations: implications for ClC-5 channel trafficking and internalization". Human Genetics. 117 (2–3): 228–37. doi:10.1007/s00439-005-1303-2. PMID 15895257.
- Piwon N, Günther W, Schwake M, Bösl MR, Jentsch TJ (November 2000). "ClC-5 Cl- -channel disruption impairs endocytosis in a mouse model for Dent's disease". Nature. 408 (6810): 369–73. doi:10.1038/35042597. PMID 11099045.
- Günther W, Piwon N, Jentsch TJ (January 2003). "The ClC-5 chloride channel knock-out mouse - an animal model for Dent's disease". Pflugers Archiv. 445 (4): 456–62. doi:10.1007/s00424-002-0950-6. PMID 12548389.
- Wang SS, Devuyst O, Courtoy PJ, Wang XT, Wang H, Wang Y, et al. (December 2000). "Mice lacking renal chloride channel, CLC-5, are a model for Dent's disease, a nephrolithiasis disorder associated with defective receptor-mediated endocytosis". Human Molecular Genetics. 9 (20): 2937–45. doi:10.1093/hmg/9.20.2937. PMID 11115837.
- Silva IV, Cebotaru V, Wang H, Wang XT, Wang SS, Guo G, et al. (April 2003). "The ClC-5 knockout mouse model of Dent's disease has renal hypercalciuria and increased bone turnover". Journal of Bone and Mineral Research. 18 (4): 615–23. doi:10.1359/jbmr.2003.18.4.615. PMID 12674322.
- Christensen EI, Devuyst O, Dom G, Nielsen R, Van der Smissen P, Verroust P, et al. (July 2003). "Loss of chloride channel ClC-5 impairs endocytosis by defective trafficking of megalin and cubilin in kidney proximal tubules". Proceedings of the National Academy of Sciences of the United States of America. 100 (14): 8472–7. doi:10.1073/pnas.1432873100. PMC 166253. PMID 12815097.
- Nielsen R, Courtoy PJ, Jacobsen C, Dom G, Lima WR, Jadot M, et al. (March 2007). "Endocytosis provides a major alternative pathway for lysosomal biogenesis in kidney proximal tubular cells". Proceedings of the National Academy of Sciences of the United States of America. 104 (13): 5407–12. doi:10.1073/pnas.0700330104. PMC 1838438. PMID 17369355.
- Devuyst O, Thakker RV (October 2010). "Dent's disease". Orphanet Journal of Rare Diseases. 5 (1): 28. doi:10.1186/1750-1172-5-28. PMC 2964617. PMID 20946626.
Further reading
- Igarashi T, Hayakawa H, Shiraga H, Kawato H, Yan K, Kawaguchi H, et al. (1995). "Hypercalciuria and nephrocalcinosis in patients with idiopathic low-molecular-weight proteinuria in Japan: is the disease identical to Dent's disease in United Kingdom?". Nephron. 69 (3): 242–7. doi:10.1159/000188464. PMID 7753256.
- Scheinman SJ, Pook MA, Wooding C, Pang JT, Frymoyer PA, Thakker RV (June 1993). "Mapping the gene causing X-linked recessive nephrolithiasis to Xp11.22 by linkage studies". The Journal of Clinical Investigation. 91 (6): 2351–7. doi:10.1172/JCI116467. PMC 443292. PMID 8099916.
- Lloyd SE, Pearce SH, Fisher SE, Steinmeyer K, Schwappach B, Scheinman SJ, et al. (February 1996). "A common molecular basis for three inherited kidney stone diseases". Nature. 379 (6564): 445–9. doi:10.1038/379445a0. PMID 8559248.
- Fisher SE, van Bakel I, Lloyd SE, Pearce SH, Thakker RV, Craig IW (October 1995). "Cloning and characterization of CLCN5, the human kidney chloride channel gene implicated in Dent disease (an X-linked hereditary nephrolithiasis)". Genomics. 29 (3): 598–606. doi:10.1006/geno.1995.9960. hdl:11858/00-001M-0000-0012-CC06-6. PMID 8575751.
- Lloyd SE, Pearce SH, Günther W, Kawaguchi H, Igarashi T, Jentsch TJ, Thakker RV (March 1997). "Idiopathic low molecular weight proteinuria associated with hypercalciuric nephrocalcinosis in Japanese children is due to mutations of the renal chloride channel (CLCN5)". The Journal of Clinical Investigation. 99 (5): 967–74. doi:10.1172/JCI119262. PMC 507905. PMID 9062355.
- Pirozzi G, McConnell SJ, Uveges AJ, Carter JM, Sparks AB, Kay BK, Fowlkes DM (June 1997). "Identification of novel human WW domain-containing proteins by cloning of ligand targets". The Journal of Biological Chemistry. 272 (23): 14611–6. doi:10.1074/jbc.272.23.14611. PMID 9169421.
- Oudet C, Martin-Coignard D, Pannetier S, Praud E, Champion G, Hanauer A (June 1997). "A second family with XLRH displays the mutation S244L in the CLCN5 gene". Human Genetics. 99 (6): 781–4. doi:10.1007/s004390050448. PMID 9187673.
- Lloyd SE, Gunther W, Pearce SH, Thomson A, Bianchi ML, Bosio M, et al. (August 1997). "Characterisation of renal chloride channel, CLCN5, mutations in hypercalciuric nephrolithiasis (kidney stones) disorders". Human Molecular Genetics. 6 (8): 1233–9. doi:10.1093/hmg/6.8.1233. PMID 9259268.
- Schurman SJ, Norden AG, Scheinman SJ (May 1998). "X-linked recessive nephrolithiasis: presentation and diagnosis in children". The Journal of Pediatrics. 132 (5): 859–62. doi:10.1016/S0022-3476(98)70318-X. PMID 9602200.
- Günther W, Lüchow A, Cluzeaud F, Vandewalle A, Jentsch TJ (July 1998). "ClC-5, the chloride channel mutated in Dent's disease, colocalizes with the proton pump in endocytotically active kidney cells". Proceedings of the National Academy of Sciences of the United States of America. 95 (14): 8075–80. doi:10.1073/pnas.95.14.8075. PMC 20931. PMID 9653142.
- Devuyst O, Christie PT, Courtoy PJ, Beauwens R, Thakker RV (February 1999). "Intra-renal and subcellular distribution of the human chloride channel, CLC-5, reveals a pathophysiological basis for Dent's disease". Human Molecular Genetics. 8 (2): 247–57. doi:10.1093/hmg/8.2.247. PMID 9931332.
- Lamb FS, Clayton GH, Liu BX, Smith RL, Barna TJ, Schutte BC (March 1999). "Expression of CLCN voltage-gated chloride channel genes in human blood vessels". Journal of Molecular and Cellular Cardiology. 31 (3): 657–66. doi:10.1006/jmcc.1998.0901. PMID 10198195.
- Strausberg RL, Feingold EA, Grouse LH, Derge JG, Klausner RD, Collins FS, et al. (December 2002). "Generation and initial analysis of more than 15,000 full-length human and mouse cDNA sequences". Proceedings of the National Academy of Sciences of the United States of America. 99 (26): 16899–903. doi:10.1073/pnas.242603899. PMC 139241. PMID 12477932.
- Moulin P, Igarashi T, Van der Smissen P, Cosyns JP, Verroust P, Thakker RV, et al. (April 2003). "Altered polarity and expression of H+-ATPase without ultrastructural changes in kidneys of Dent's disease patients". Kidney International. 63 (4): 1285–95. doi:10.1046/j.1523-1755.2003.00851.x. PMID 12631345.
- Wu F, Roche P, Christie PT, Loh NY, Reed AA, Esnouf RM, Thakker RV (April 2003). "Modeling study of human renal chloride channel (hCLC-5) mutations suggests a structural-functional relationship". Kidney International. 63 (4): 1426–32. doi:10.1046/j.1523-1755.2003.00859.x. PMID 12631358.
- Carballo-Trujillo I, Garcia-Nieto V, Moya-Angeler FJ, Antón-Gamero M, Loris C, Méndez-Alvarez S, Claverie-Martin F (April 2003). "Novel truncating mutations in the ClC-5 chloride channel gene in patients with Dent's disease". Nephrology, Dialysis, Transplantation. 18 (4): 717–23. doi:10.1093/ndt/gfg016. PMID 12637640.
- Ludwig M, Waldegger S, Nuutinen M, Bökenkamp A, Reissinger A, Steckelbroeck S, Utsch B (2004). "Four additional CLCN5 exons encode a widely expressed novel long CLC-5 isoform but fail to explain Dent's phenotype in patients without mutations in the short variant". Kidney & Blood Pressure Research. 26 (3): 176–84. doi:10.1159/000071883. PMID 12886045.
- Hryciw DH, Wang Y, Devuyst O, Pollock CA, Poronnik P, Guggino WB (October 2003). "Cofilin interacts with ClC-5 and regulates albumin uptake in proximal tubule cell lines" (PDF). The Journal of Biological Chemistry. 278 (41): 40169–76. doi:10.1074/jbc.M307890200. PMID 12904289.
External links
- CLCN5+protein,+human at the US National Library of Medicine Medical Subject Headings (MeSH)
- Human CLCN5 genome location and CLCN5 gene details page in the UCSC Genome Browser.
This article incorporates text from the United States National Library of Medicine, which is in the public domain.
PDB gallery | |
|---|---|
|