Antagonistic pleiotropy hypothesis
The antagonistic pleiotropy hypothesis was first proposed by George C. Williams in 1957 as an evolutionary explanation for senescence.[1] Pleiotropy is the phenomenon where one gene controls for more than one phenotypic trait in an organism.[2] Antagonistic pleiotropy is when one gene controls for more than one trait, where at least one of these traits is beneficial to the organism's fitness early on in life and at least one is detrimental to the organism's fitness later on due to a decline in the force of natural selection.[3][4] The theme of G.C. William's idea about antagonistic pleiotropy was that if a gene caused both increased reproduction in early life and aging in later life, then senescence would be adaptive in evolution. For example, one study suggests that since follicular depletion in human females causes both more regular cycles in early life and loss of fertility later in life through menopause, it can be selected for by having its early benefits outweigh its late costs.[5]
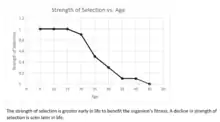
As a constraint on perfection
Antagonistic pleiotropy is one of the several reasons evolutionary biologists give for organisms never being able to reach perfection through natural selection. Antagonistically pleiotropic genes are the explanation for fitness trade-offs.[3] This means that genes that are pleiotropic control for some beneficial traits and some detrimental traits; thus, if they happen to persist through natural selection, this will prevent organisms from reaching perfection because if they possess the benefits of the gene, they must also possess the imperfections or faults. An example of this would be female rodents that live in a nest with other females and may end up feeding young that are not theirs due to their intense parental drive.[6] This strong parental drive will be selected for, but the organisms will still make the mistake of feeding young that are not theirs and misallocating their resources.
Benefits and costs
Antagonistic pleiotropy has several negative consequences. It results in delayed adaptation, an altered path of evolution, and reduced adaptation of other traits.[7] In addition, the overall benefit of alleles is cut down significantly (by about half) by pleiotropy. Still, antagonistic pleiotropy has some evolutionary benefits. In fact, the conservation of genes is directly related to the pleiotropic character of an organism.[8] This implies that genes that control for multiple traits, even if the traits have different implications for the organism's fitness, have more staying power in an evolutionary context.
Role in sexual selection
It is generally accepted that the evolution of secondary sexual characteristics persists until the relative costs of survival outweigh the benefits of reproductive success.[9] At the level of genes, this means a trade-off between variation and expression of selected traits. Strong, persistent sexual selection should result in decreased genetic variation for these traits. However, higher levels of variation have been reported in sexually-selected traits compared to non-sexually selected traits.[10] This phenomenon is especially clear in lek species, where males confer no immediate advantage to the female. Female choice presumably depends on correlating male displays (secondary sexual characteristics) with overall genetic quality. If such directional sexual selection depletes variation in males, why would female choice continue to exist? Rowe and Houle answer this question (the lek paradox) using the notion of genetic capture, which couples the sexually-selected traits with the overall condition of the organism. They posit that the genes for secondary sexual characteristics must be pleiotropically linked to condition, a measure of the organism's fitness. In other words, the genetic variation in secondary sexual characteristics is maintained due to variation in the organism's condition.[11]
Role in disease
The survival of many serious genetic disorders in our long evolutionary history has led researchers to reassess the role of antagonistic pleiotropy in disease. If genetic disorders are defined by the existence of deleterious alleles, then natural selection acting over evolutionary time would result in a lower frequency of mutations than are currently observed.[12] In a recent article, Carter and Nguyen identify several genetic disorders, arguing that far from being a rare phenomenon, antagonistic pleiotropy might be a fundamental mechanism for the survival of these non-optimal alleles.
In one of these studies, 99 individuals with Laron syndrome (a rare form of dwarfism) were monitored alongside their non-dwarf kin for a period of ten years. Patients with Laron syndrome possess one of three genotypes for the growth hormone receptor gene (GHR). Most patients have an A->G splice site mutation in position 180 in exon 6. Some others possess a nonsense mutation (R43X), while the rest are heterozygous for the two mutations. Laron syndrome patients experienced a lower incidence of cancer mortality and diabetes compared to their non-dwarf kin.[13] This suggests a role for antagonistic pleiotropy, whereby a deleterious mutation is preserved in a population because it still confers some survival benefit.
Another instance of antagonistic pleiotropy is manifested in Huntington's disease, a rare neurodegenerative disorder characterized by a high number of CAG repeats within the Huntingtin gene. The onset of Huntington's is usually observed post-reproductive age and generally involves involuntary muscle spasms, cognitive difficulties and psychiatric problems. Incidentally, the high number of CAG repeats is associated with increased activity of p53, a tumor suppressing protein that participates in apoptosis. It has been hypothesized that this explains the lower rates of cancer among Huntington's patients. Huntington's disease is also correlated with high fecundity.[12]
Additionally, it was found that individuals with a higher pro-inflammatory ratio TNFα/IL-10 had a significantly higher incidence of death due to cardiovascular disease in old age. Yet, it was hypothesized that this genotype was prevalent because higher ratios of TNFα/IL-10 allow individuals to more effectively combat infection during reproductive years.[14]
Sickle cell anemia, Beta-thalassemia, and cystic fibrosis are some other examples of the role antagonistic pleiotropy may play in genetic disorders.[12]
Ubiquity
Although there are so many negative effects related to genes that are antagonistically pleiotropic, it is still present among most forms of life. Indeed, pleiotropy is one of the most common traits possessed by genes overall.[8] In addition to that, pleiotropy is under strong stabilizing selection.[7] In one experiment with mice and the morphology of the mandible, 1/5 of the loci had effects of pleiotropy for the entire mandible.[2] One other example was in the Russian biologist Dmitry K. Belyaev's study on the domestication of the fox.[15] In Dmitry K. Belyaev's farm-fox experiment, wild foxes were bred for docile behavior alone. After 40 generations, other physiological changes had surfaced including shortened tails, floppy ears, a white star in the forehead, rolled tails, shorter legs. Since the only thing being selected for was behavior, this leads scientists to believe that these secondary characteristics were controlled by the same gene or genes as docile behavior.
Adaptivity and senescence
An antagonistically pleiotropic gene can be selected for if it has beneficial effects in early life while having its negative effects in later life because genes tend to have larger impacts on fitness in an organism's prime than in their old age.[5] An example of this is testosterone levels in male humans. Higher levels of this hormone lead to increased fitness in early life, while causing decreased fitness in later life due to a higher risk for prostate cancer.[16] This is an example of antagonistic pleiotropy being an explanation for senescence. Senescence is the act of ageing in individuals; it's the failure over time of the individual's life processes by natural causes.[17] Williams's theory has been the motivation for many of the experimental studies on the reasons for aging in the last 25 years.[18] However, there is more than one theory out there for aging. The competing model to explain senescence is Medawar's "mutation accumulation" hypothesis, saying that "over evolutionary time, late-acting mutations will accumulate at a much faster rate than early-acting mutation. These late-acting mutations will thus lead to declining viability and/or fertility as an organism ages."[18] Medawar's theory is based around the older concept of selection shadow that had been discussed throughout the early 1900s and led to Medawar's theory after discussions with J. B. S. Haldane in the 1940s.[19]
Potential Examples
DNA Damage Theory of Aging
A prominent explanation for aging at the molecular level is the DNA damage theory of aging. It has been proposed that genetic elements that regulate DNA repair in somatic cells may constitute an important example of age-dependent pleiotropic "genes".[20][21] As pointed out by Vijg,[21] genome repair and maintenance is beneficial early in life by swiftly eliminating DNA damage or damaged cells. However, studies of DNA repair in the brain[22][23] and in muscle[24] indicate that the transition from mitotic cell division to the post-mitotic condition that occurs early in life is accompanied by a reduction in DNA repair. The reduced expression of DNA repair is presumably part of an evolutionary adaptation for diverting the resources of the cell that were previously used for DNA repair, as well as for replication and cell division, to more essential neuronal and muscular functions.[20]
The harmful effect of this genetically controlled reduction in expression is to allow increased accumulation of DNA damage. Reduced DNA repair causes increased impairment of transcription and progressive loss of cell and tissue function. However, these harmful effects of DNA damage are cumulative and most severe in chronologically older individuals whose numbers diminish with time (by causes of death that can be independent of senescence). As a consequence, the beneficial effects of the genetic elements that control the reduction of DNA repair early in life would predominate. Thus regulatory genetic elements that reduce expression of DNA repair genes in post-mitotic cells appear to be important examples of the postulated pleiotropic "genes" that are beneficial in youth but deleterious at an older age.[20]
Telomere Theory
Another example related to aging is the Telomere theory. Telomere theory proposes that telomeres shorten with repeated cell division which attribute to cell senescence and tissue damage.[25] The end replication problem explains the mechanism behind the inability of DNA polymerase to commence the RNA primer to perform its function in completing the lagging strand due to the shortening of DNA. Telomere shortening is common in somatic cells. However, germ line and stem cells prevent the end replication problem with the help of telomerase. Telomerase elongates the 3’ end that is then formed into a t-loop to prevent the cell from entering the G0 phase and cell senescence.[25]
Inflammation and damage to tissue are the underlying problems due to increased senescent cells. In several studies shortened telomeres have been associated with age related sarcopenia,[26] atherosclerotic cardiovascular disease,[27] and cancer.[28] However, there is still the question whether telomere length causes these diseases or if the diseases cause shortened telomeres. Hence, the shortening of telomeres complies with antagonistic pleiotropy theory. The trade-off exists as the cell benefits from telomerase which prevents permanent growth arrest but telomere shortening is associated with functional loss.
Free Radical Theory
Another example related to aging is the Free Radical theory. Free Radical Theory suggests that the free radicals, which are being produced by aerobic respiration, are causing oxidative stress to be put on the body. This oxidative stress will result in aging and lead to death.[29] Oxygen centered radicals are very reactive and can cause the accumulation of damage on lipids, nucleic acids as well as proteins within the body. This accumulation of damage on the biological molecules changes the framework and leads to a reduction in the molecules' activity levels. Lipid peroxides accumulate in the membrane phospholipids, which in turn diminishes the mitochondrial membrane's effectiveness as a barrier. The process of DNA transcription and translation also acquires oxidative damage. The result is alterations in the base pairings of the DNA sequence. Research has found that DNA mutations from free radical damage are highly uncommon but would still lead to the build up of damaged proteins as well as decreased biological activity.[25]
See also
References
- Williams G.C. (1957). "Pleiotropy, natural selection, and the evolution of senescence". Evolution. 11 (4): 398–411. doi:10.2307/2406060. JSTOR 2406060.
- Cheverud J (1996). "Developmental integration and the evolution of pleiotropy". American Zoology. 36: 44–50. CiteSeerX 10.1.1.526.8366. doi:10.1093/icb/36.1.44.
- Elena S.F.; Sanjuán R. (2003). "Climb every mountain?". Science. 302 (5653): 2074–2075. doi:10.1126/science.1093165. PMID 14684807. S2CID 83853360.
- Flatt, Thomas (2011-05-05). "Survival costs of reproduction in Drosophila" (PDF). Experimental Gerontology. 46 (5): 369–375. doi:10.1016/j.exger.2010.10.008. ISSN 0531-5565. PMID 20970491. S2CID 107465469.
- Wood, J.W., K.A. O'Conner, D.J. Holman, E. Bringle, S.H. Barsom, M.A. Grimes. 2001. The evolution of menopause by antagonistic pleiotropy. Center for Demography and Ecology, Working Paper.
- Alcock, J. 2005. Animal Behavior: eighth edition
- Otto S.P. (2004). "Two steps forward, one step back: the pleiotropic effects of favoured alleles". Proc. Biol. Sci. 271 (1540): 705–714. doi:10.1098/rspb.2003.2635. PMC 1691650. PMID 15209104.
- He X.; Zhang J. (2006). "Toward a Molecular Understanding of Pleiotropy". Genetics. 173 (4): 1885–1891. doi:10.1534/genetics.106.060269. PMC 1569710. PMID 16702416.
- Bolstad, Geir; Pelabon, Cristophe; Larsen, Line-K; Fleming, Ian A.; Viken, Aslaug; Rosenqvist, Gunilla (2012). "The Effect of Purging on Sexually Selected Traits through Antagonistic Pleiotropy with Survival". Ecology and Evolution. 2 (6): 1184–191. doi:10.1002/ece3.246. PMC 3402193. PMID 22833793.
- Pomiankowski, A; AP Moller (1995). "A Resolution of the Lek Paradox". Proceedings of the Royal Society of London. 260 (1357): 21–29. doi:10.1098/rspb.1995.0054. S2CID 43984154.
- Rowe, L; D. Houle (1996). "The Lek Paradox and the Capture of Genetic Variance by Condition Dependent Traits". Proceedings of the Royal Society of London. 263 (1375): 1415–1421. Bibcode:1996RSPSB.263.1415R. doi:10.1098/rspb.1996.0207. S2CID 85631446.
- Carter, Ashley; Andrew Q. Nguyen (2011). "Antagonistic Pleiotropy as a Widespread Mechanism for the Maintenance of Polymorphic Disease Alleles". BMC Medical Genetics. 12: 160. doi:10.1186/1471-2350-12-160. PMC 3254080. PMID 22151998.
- Guevara-Aguirre, J.; Balasubramanian, P.; Guevara-Aguirre, M.; Wei, M.; Madia, F.; Chang, C.W.; Hwang, D. (2011). "Growth Hormone Receptor Deficiency is Associated with a Major Reduction in Pro-aging Signaling, Cancer, and Diabetes in Humans". Science Translational Medicine. 3 (70): 70ra13. doi:10.1126/scitranslmed.3001845. PMC 3357623. PMID 21325617.
- Van Den Biggelaar, AH (2004). "Inflammation underlying cardiovascular mortality is a late consequence of evolutionary programming". The FASEB Journal. 12 (9): 1022–1024. doi:10.1096/fj.03-1162fje. PMID 15084512.
- Trut L.N. (1996). "Early canid domestication: the farm-fox experiment". American Scientist. 87 (2): 160. Bibcode:1999AmSci..87.....T. doi:10.1511/1999.2.160.
- Gann P.H.; Hennekens C.H.; Ma J.; Longcope C.; Stampfer M.J. (1996). "Prospective Study of Sex Hormone Levels and Risk of Prostate Cancer". Journal of the National Cancer Institute. 88 (16): 1118–1126. doi:10.1093/jnci/88.16.1118. PMID 8757191.
- Promislow D.E.L. (2004). "Protein networks, pleiotropy and the evolution of senescence". Proc. Biol. Sci. 271 (1545): 1225–1234. doi:10.1098/rspb.2004.2732. PMC 1691725. PMID 15306346.
- Fox, C.W. and J.B. Wolf. 2006. Evolutionary Genetics: Concepts and Case Studies.
- Fabian, Daniel; Flatt, Thomas (2011). "The Evolution of Aging". Scitable. Nature Publishing Group. Retrieved May 20, 2014.
- Bernstein C, Bernstein H. (1991) Aging, Sex, and DNA Repair. Academic Press, San Diego. ISBN 0120928604 ISBN 978-0120928606
- Vijg J (2014). "Aging genomes: a necessary evil in the logic of life". BioEssays. 36 (3): 282–92. doi:10.1002/bies.201300127. PMC 5985526. PMID 24464418.
- Gensler HL (1981). "Low level of U.V.-induced unscheduled DNA synthesis in postmitotic brain cells of hamsters: possible relevance to aging". Exp. Gerontol. 16 (2): 199–207. doi:10.1016/0531-5565(81)90046-2. PMID 7286098. S2CID 6261990.
- Karran P, Moscona A, Strauss B (1977). "Developmental decline in DNA repair in neural retina cells of chick embryos. Persistent deficiency of repair competence in a cell line derived from late embryos". J. Cell Biol. 74 (1): 274–86. doi:10.1083/jcb.74.1.274. PMC 2109876. PMID 559680.
- Lampidis TJ, Schaiberger GE (1975). "Age-related loss of DNA repair synthesis in isolated rat myocardial cells". Exp. Cell Res. 96 (2): 412–6. doi:10.1016/0014-4827(75)90276-1. PMID 1193184.
- McDonald, Roger B. (2019-06-07), "Basic Concepts in the Biology of Aging", Biology of Aging, Garland Science, pp. 1–36, doi:10.1201/9780429030642-1, ISBN 978-0-429-03064-2
- Araújo Carvalho, Aline Carla; Tavares Mendes, Mário Luis; da Silva Reis, Monique Carla; Santos, Victor Santana; Tanajura, Diego Moura; Martins-Filho, Paulo Ricardo Saquete (September 2019). "Telomere length and frailty in older adults—A systematic review and meta-analysis". Ageing Research Reviews. 54: 100914. doi:10.1016/j.arr.2019.100914. PMID 31170457. S2CID 173992082.
- De Meyer, Tim; Nawrot, Tim; Bekaert, Sofie; De Buyzere, Marc L.; Rietzschel, Ernst R.; Andrés, Vicente (August 2018). "Telomere Length as Cardiovascular Aging Biomarker". Journal of the American College of Cardiology. 72 (7): 805–813. doi:10.1016/j.jacc.2018.06.014. PMID 30092957.
- Pepper, Chris; Norris, Kevin; Fegan, Christopher (February 2020). "Clinical utility of telomere length measurements in cancer". Current Opinion in Genetics & Development. 60: 107–111. doi:10.1016/j.gde.2020.02.012. PMID 32220800.
- Beckman, Kenneth B.; Ames, Bruce N. (1998-04-01). "The Free Radical Theory of Aging Matures". Physiological Reviews. 78 (2): 547–581. doi:10.1152/physrev.1998.78.2.547. ISSN 0031-9333. PMID 9562038.