Carbon–fluorine bond
The carbon–fluorine bond is a polar covalent bond between carbon and fluorine that is a component of all organofluorine compounds. It is one of the strongest single bonds in organic chemistry—behind the B-F single bond, Si-F single bond and the H-F single bond, and relatively short—due to its partial ionic character. The bond also strengthens and shortens as more fluorines are added to the same carbon on a chemical compound. As such, fluoroalkanes like tetrafluoromethane (carbon tetrafluoride) are some of the most unreactive organic compounds.
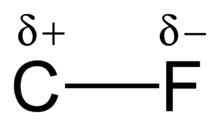
Electronegativity and bond strength
The high electronegativity of fluorine (4.0 for fluorine vs. 2.5 for carbon) gives the carbon–fluorine bond a significant polarity/dipole moment. The electron density is concentrated around the fluorine, leaving the carbon relatively electron poor. This introduces ionic character to the bond through partial charges (Cδ+—Fδ−). The partial charges on the fluorine and carbon are attractive, contributing to the unusual bond strength of the carbon–fluorine bond. The bond is labeled as "the strongest in organic chemistry,"[1] because fluorine forms the strongest single bond to carbon. Carbon–fluorine bonds can have a bond dissociation energy (BDE) of up to 130 kcal/mol.[2] The BDE (strength of the bond) of C-F is higher than other carbon–halogen and carbon–hydrogen bonds. For example, the BDEs of the C-X bond within a CH3-X molecule is 115, 104.9, 83.7, 72.1, and 57.6 kcal/mol for X = fluorine, hydrogen, chlorine, bromine, and iodine, respectively.[3]
Bond length
The carbon–fluorine bond length is typically about 1.35 ångström (1.39 Å in fluoromethane).[1] It is shorter than any other carbon–halogen bond, and shorter than single carbon–nitrogen and carbon–oxygen bonds, despite fluorine having a larger atomic mass. The short length of the bond can also be attributed to the ionic character/electrostatic attractions between the partial charges on carbon and fluorine. The carbon–fluorine bond length varies by several hundredths of an ångstrom depending on the hybridization of the carbon atom and the presence of other substituents on the carbon or even in atoms farther away. These fluctuations can be used as indication of subtle hybridization changes and stereoelectronic interactions. The table below shows how the average bond length varies in different bonding environments (carbon atoms are sp3-hybridized unless otherwise indicated for sp2 or aromatic carbon).
Bond Mean bond length (Å)[4] CCH2F, C2CHF 1.399 C3CF 1.428 C2CF2, H2CF2, CCHF2 1.349 CCF3 1.346 FCNO2 1.320 FCCF 1.371 Csp2F 1.340 CarF 1.363 FCarCarF 1.340
The variability in bond lengths and the shortening of bonds to fluorine due to their partial ionic character are also observed for bonds between fluorine and other elements, and have been a source of difficulties with the selection of an appropriate value for the covalent radius of fluorine. Linus Pauling originally suggested 64 pm, but that value was eventually replaced by 72 pm, which is half of the fluorine–fluorine bond length. However, 72 pm is too long to be representative of the lengths of the bonds between fluorine and other elements, so values between 54 pm and 60 pm have been suggested by other authors.[5][6][7][8]
Bond strength effect of geminal bonds
With increasing number of fluorine atoms on the same (geminal) carbon the other bonds become stronger and shorter. This can be seen by the changes in bond length and strength (BDE) for the fluoromethane series, as shown on the table below; also, the partial charges (qC and qF) on the atoms change within the series.[2] The partial charge on carbon becomes more positive as fluorines are added, increasing the electrostatic interactions, and ionic character, between the fluorines and carbon.
Compound C-F bond length (Å) BDE (kcal/mol) qC qF CH3F 1.385 109.9 ± 1 0.01 −0.23 CH2F2 1.357 119.5 0.40 −0.23 CHF3 1.332 127.5 0.56 −0.21 CF4 1.319 130.5 ± 3 0.72 −0.18
Gauche effect
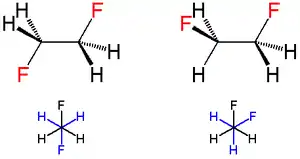
When two fluorine atoms are in vicinal (i.e., adjacent) carbons, as in 1,2-difluoroethane (H2FCCFH2), the gauche conformer is more stable than the anti conformer—this is the opposite of what would normally be expected and to what is observed for most 1,2-disubstituted ethanes; this phenomenon is known as the gauche effect.[9] In 1,2-difluoroethane, the gauche conformation is more stable than the anti conformation by 2.4 to 3.4 kJ/mole in the gas phase. This effect is not unique to the halogen fluorine, however; the gauche effect is also observed for 1,2-dimethoxyethane. A related effect is the alkene cis effect. For instance, the cis isomer of 1,2-difluoroethylene is more stable than the trans isomer.[10]
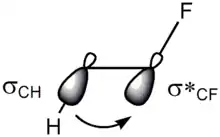
There are two main explanations for the gauche effect: hyperconjugation and bent bonds. In the hyperconjugation model, the donation of electron density from the carbon–hydrogen σ bonding orbital to the carbon–fluorine σ* antibonding orbital is considered the source of stabilization in the gauche isomer. Due to the greater electronegativity of fluorine, the carbon–hydrogen σ orbital is a better electron donor than the carbon–fluorine σ orbital, while the carbon–fluorine σ* orbital is a better electron acceptor than the carbon–hydrogen σ* orbital. Only the gauche conformation allows good overlap between the better donor and the better acceptor.[11]
Key in the bent bond explanation of the gauche effect in difluoroethane is the increased p orbital character of both carbon–fluorine bonds due to the large electronegativity of fluorine. As a result, electron density builds up above and below to the left and right of the central carbon–carbon bond. The resulting reduced orbital overlap can be partially compensated when a gauche conformation is assumed, forming a bent bond. Of these two models, hyperconjugation is generally considered the principal cause behind the gauche effect in difluoroethane.[1][12]
Spectroscopy
The carbon–fluorine bond stretching appears in the infrared spectrum between 1000 and 1360 cm−1. The wide range is due to the sensitivity of the stretching frequency to other substituents in the molecule. Monofluorinated compounds have a strong band between 1000 and 1110 cm−1; with more than one fluorine atoms, the band splits into two bands, one for the symmetric mode and one for the asymmetric.[13] The carbon–fluorine bands are so strong that they may obscure any carbon–hydrogen bands that might be present.[14]
Organofluorine compounds can also be characterized using NMR spectroscopy, using carbon-13, fluorine-19 (the only natural fluorine isotope), or hydrogen-1 (if present). The chemical shifts in 19F NMR appear over a very wide range, depending on the degree of substitution and functional group. The table below shows the ranges for some of the major classes.[15]
Type of Compound Chemical Shift Range (ppm) Relative to neat CFCl3 F–C=O −70 to −20 CF3 +40 to +80 CF2 +80 to +140 CF +140 to +250 ArF +80 to +170
See also
References
- O'Hagan D (February 2008). "Understanding organofluorine chemistry. An introduction to the C–F bond". Chem Soc Rev. 37 (2): 308–19. doi:10.1039/b711844a. PMID 18197347.
- Lemal DM. "Perspective on Fluorocarbon Chemistry" J Org Chem. 2004, volume 69, p 1–11. doi:10.1021/jo0302556
- Blanksby SJ, Ellison GB (April 2003). "Bond dissociation energies of organic molecules". Acc. Chem. Res. 36 (4): 255–63. CiteSeerX 10.1.1.616.3043. doi:10.1021/ar020230d. PMID 12693923.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen. Tables of bond Lengths determined by X-Ray and Neutron Diffraction. Part 1. Bond Lengths in Organic Compounds. J. Chem. Soc. Perkin Trans. II 1987, S1-S19.
- Gillespie Ronald; Robinson Edward (1992). "Bond Lengths in Covalent Fluorides. A New Value for the Covalent Radius of Fluorine". Inorganic Chemistry. 31 (10): 1960–1963. doi:10.1021/ic00036a045.
- Robinson Edward; Johnson Samuel; Tang Ting-Hua; Gillespie Ronald (1997). "Reinterpretation of the Lengths of Bonds to Fluorine in Terms of an Almost Ionic Model". Inorganic Chemistry. 36 (14): 3022–3030. doi:10.1021/ic961315b. PMID 11669953.
- Cordero Beatriz; Gómez Verónica; Platero-Prats Ana E; Revés Marc; Echeverría Jorge; Cremades Eduard; Barragán Flavia; Alvarez Santiago (2008). "Covalent radii revisited". Dalton Trans. 2008 (21): 2832–2838. doi:10.1039/b801115j. PMID 18478144. S2CID 244110.
- Pyykkö P.; Atsumi M. (2009). "Molecular Single-Bond Covalent Radii for Elements 1-118". Chemistry: A European Journal. 15 (1): 186–197. doi:10.1002/chem.200800987. PMID 19058281.
- Contribution to the Study of the Gauche Effect. The Complete Structure of the Anti Rotamer of 1,2-Difluoroethane Norman C. Craig, Anthony Chen, Ki Hwan Suh, Stefan Klee, Georg C. Mellau, Brenda P. Winnewisser, and Manfred Winnewisser J. Am. Chem. Soc.; 1997; 119(20) pp 4789 - 4790; (Communication) doi:10.1021/ja963819e
- The stereochemical consequences of electron delocalization in extended .pi. systems. An interpretation of the cis effect exhibited by 1,2-disubstituted ethylenes and related phenomena Richard C. Bingham J. Am. Chem. Soc.; 1976; 98(2); 535-540 Abstract
- Alabugin, I. V. Stereoelectronic Effects: the Bridge between Structure and Reactivity. John Wiley & Sons Ltd, Chichester, UK, 2016
- Goodman, L.; Gu, H.; Pophristic, V.. Gauche Effect in 1,2-Difluoroethane. Hyperconjugation, Bent Bonds, Steric Repulsion. J. Phys. Chem. A. 2005, 109, 1223-1229. doi:10.1021/jp046290d
- George Socrates; Socrates (2001). Infrared and Raman characteristic group frequencies: tables and charts. John Wiley and Sons. p. 198. ISBN 978-0-470-09307-8.
- Barbara H. Stuart (2004). Infrared Spectroscopy: Fundamentals and Applications. John Wiley and Sons. p. 82. ISBN 978-0-470-85428-0.
- "Archived copy". Archived from the original on 2008-05-15. Retrieved 2008-11-09.CS1 maint: archived copy as title (link)
Compounds of carbon with other elements in the periodic table | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Legend |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||