Organoboron chemistry
Organoborane or organoboron compounds are chemical compounds of boron and carbon that are organic derivatives of BH3, for example trialkyl boranes. Organoboron chemistry or organoborane chemistry is the chemistry of these compounds.[1][2]

Organoboron compounds are important reagents in organic chemistry enabling many chemical transformations, the most important one called hydroboration. Reactions of organoborates and boranes involve the transfer of a nucleophilic group attached to boron to an electrophilic center either inter- or intramolecularly. α,β-Usaturated borates, as well as borates with a leaving group at the α position, are highly susceptible to intramolecular 1,2-migration of a group from boron to the electrophilic α position. Oxidation or protonolysis of the resulting organoboranes may generate a variety of organic products, including alcohols, carbonyl compounds, alkenes, and halides.[3]
Properties of the B-C bond
The C-B bond has low polarity (the difference in electronegativity 2.55 for carbon and 2.04 for boron), and therefore alkyl boron compounds are in general stable though easily oxidized.
In part because its lower electronegativity, boron often forms electron-deficient compounds, such as the triorganoboranes. Vinyl groups and aryl groups donate electrons and make boron less electrophilic and the C-B bond gains some double bond character. Like the parent borane, diborane, organoboranes are classified in organic chemistry as strong electrophiles because boron is unable to gain a full octet of electrons. Unlike diborane however, most organoboranes do not form dimers.
Synthesis
From Grignard reagents
Simple organoboranes such as triethylborane or tris(pentafluorophenyl)boron can be prepared from trifluoroborane (as the ether complex) and the ethyl or pentafluorophenyl Grignard reagent. The borates (R4B−) are generated via addition of R−-equivalents (RMgX, RLi, etc.) to R3B.
From alkenes
Alkenes insert into B-H bonds of boranes in a process called hydroboration. The process involves anti-Markovnikov addition. Hydroboration of alkenes or alkynes with borane (BH3) or borane equivalents leads to the conversion of only 33% of the starting olefin to product after oxidation or protonolysis—the remaining olefin is incorporated into boron-containing byproducts. One organoboron reagent that is often employed in synthesis is 9-BBN.[4] Hydroborations take place stereospecifically in a syn mode, that is on the same face of the alkene. In this concerted reaction the transition state is represented as a square with the corners occupied by carbon, carbon, hydrogen and boron with maximum overlap between the two olefin p-orbitals and the empty boron orbital.
By borylation
Metal-catalyzed C-H Borylation reactions are transition metal catalyzed organic reactions that produce an organoboron compound through functionalization of aliphatic and aromatic C-H bonds. A common reagent in this type of reaction is bis(pinacolato)diboron.
Classes of organoboron compounds
Organoboranes and hydrides
Among the most studied classes of organoboron compounds have the formula BRnH3−n. As discussed above, these compounds are used as catalysts, reagents, and synthetic intermediates. The trialkyl and triaryl derivatives feature trigonal planar boron center that is typically only weakly Lewis acidic. Except for a few very bulky derivatives, the hydrides (BRnH3−n for n = 1 or 2) exist as dimers, reminiscent of the structure of diborane itself. Trisubstituted derivatives, e.g. triethylboron are monomers.[5]
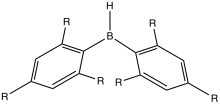
Borinic and boronic acids and esters (BRn(OR)3-n)
Compounds of the type BRn(OR)3-n are called borinic esters (n = 2), boronic esters (n = 1), and borates (n = 0). Boronic acids are used in Suzuki reaction. Trimethyl borate, which is debatably not an organoboron compound, is an intermediate in the production of sodium borohydride.
Boron clusters
Boron is renowned for forming cluster compounds, e.g. dodecaborate [B12H12]2-. Many organic derivatives are known for such clusters. One example is [B12(CH3)12]2- and its radical derivative [B12(CH3)12]−.[7] Related cluster compounds with carbon vertices are called carboranes. The best known is orthocarborane, with the formula C2B10H12. Although they have few commercial applications, carboranes have attracted much attention because they are so structurally unusual. Anionic derivatives, dicarbollides, e.g., [C2B9H11]2− are ligands that behave like cyclopentadienide.
Bora-substituted aromatic compounds
In borabenzene, one CH center in benzene is replaced by boron. These compounds are invariably isolated as adducts, e.g., C5H5B-pyridine. The cyclic compound borole, a structural analog of pyrrole, has not been isolated, but substituted derivatives known as boroles are known. The cyclic compound borepin is aromatic.
Boryl compounds
Boryl anions have the formula R2B−. Nucleophilic anionic boryl compounds have long been elusive but a 2006 study described a boryllithium compound, which reacts as a nucleophile:[8][9] Organometallic compounds with metal to boron bonds, (i.e., M–BR2), are known as boryl complexes. Related ligands are borylenes (M–B(R)–M).
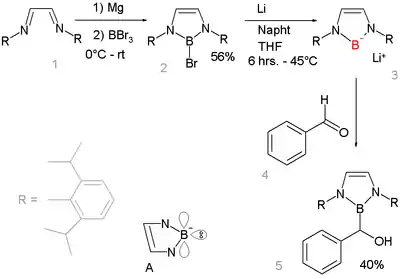
The absence of lithium boryl compounds is notable because in other period 2 elements lithium salts are common e.g. lithium fluoride, lithium hydroxide, lithium amide, and methyllithium. The gap highlights the very low electronegativity of boron. Reaction of base with a borohydride R2BH does not result in deprotonation to the boryl anion R2B− but to formation of the boryl anion R2B−H(base)+. This reaction product has a complete octet.[10] Instead the boryl compound is prepared by reductive heterolysis of a boron-bromide bond by lithium metal. The new boryl lithium compound is very similar to and isoelectronic with N-heterocyclic carbenes. It is designed to benefit from aromatic stabilization (6-electron system counting the nitrogen lone pairs and an empty boron p-orbital, see structure A) and from kinetic stabilization from the bulky 2,6-diisopropylphenyl groups. X-ray diffraction confirms sp2 hybridization at boron and its nucleophilic addition reaction with benzaldehyde gives further proof of the proposed structure.
Alkylideneboranes
Alkylideneboranes of the type RB=CRR with a boron – carbon double bond are rarely encountered. An example is borabenzene. The parent compound is HB=CH2 which can be detected at low temperatures. A fairly stable derivative is CH3B=C(SiMe3)2 but is prone to cyclodimerisation.[11]
NHC adducts of boron
NHCs and boranes form stable NHC borane adducts.[12] Triethylborane adducts can be synthesised directly from the imidazolium salt and lithium triethylborohydride. Members of this compound class are investigated for use as reagent or catalyst.
Diborenes
Chemical compounds with boron to boron double bonds are rare. In 2007 the first neutral diborene (RHB=BHR) was presented by Gregory Robinson of the University of Georgia.[13][14][15] Each boron atom has a proton attached to it and each boron atom is coordinated to a NHC carbene. The parent structure with the additional carbene ligands is diborane(2).[16][17]
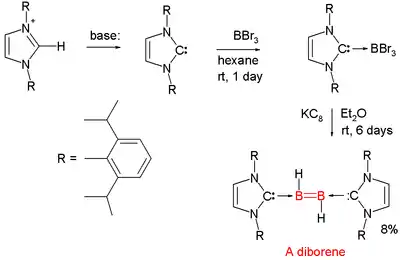
A reported diboryne is based on similar chemistry.
Reactions
Organoboranes (R3B) and borates (R4B−, generated via addition of R− to R3B) possess boron–carbon bonds that are polarized toward carbon. Thus, the carbon attached to boron is nucleophilic,[18] and in borates this property may be harnessed to transfer one of the R groups to an electrophilic center either inter- or (more often) intramolecularly.[19] In the latter case, the nucleophilic R group is able to undergo 1,2-migration towards an electrophilic carbon attached to boron.[20] The resulting reorganized borane can then be oxidized or subjected to protonolysis to afford organic products. Examples covered in this article are shown below.
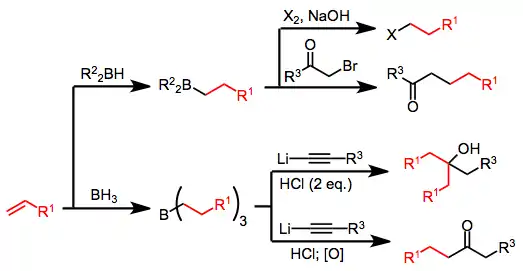
Hydroboration of alkenes or alkynes is an efficient method for the generation of boranes; however, the use of borane (BH3) or borane equivalents leads to the conversion of only 33% of the starting olefin to product after oxidation or protonolysis—the remaining olefin is incorporated into boron-containing byproducts. The use of a stoichiometric amount of 9-borabicyclo[3.3.1]nonane (9-BBN) as the hydroborating reagent provides a solution to this problem.[21]
Hydroboration-oxidation
In organic synthesis the hydroboration reaction is taken further to generate other functional groups in the place of the boron group. The hydroboration-oxidation reaction offers a route to alcohols by oxidation of the borane with hydrogen peroxide or to the carbonyl group with the stronger oxidizing agent chromium oxide.
Rearrangements
Carbon monoxide is found to react with trialkylboranes. What follows is a 1,2-rearrangement whereby an alkyl substituent migrates from boron to the carbon of the carbonyl group. Homologated primary alcohols result from the treatment of organoboranes with carbon monoxide and a hydride.[22]
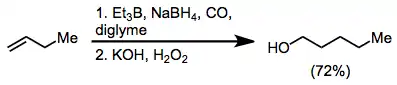
Allylboration
Asymmetric allylboration demonstrates another useful application of organoboranes in carbon–carbon bond formation.[23] In this example from Nicolaou's synthesis of the epothilones,[24] asymmetric allylboration (using an allylborane derived from chiral alpha-pinene) is used in conjunction with TBS protection and ozonolysis. Overall, this provides a two-carbon homologation sequence that delivers the required acetogenin sequence.
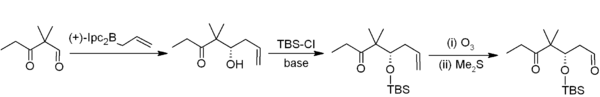
As reducing agent
Borane hydrides such as 9-BBN and L-selectride (lithium tri-sec-butylborohydride) are reducing agents. An example of an asymmetric catalyst for carbonyl reductions is the CBS catalyst. This catalyst is also based on boron, the purpose of which is coordination to the carbonyl oxygen atom.
Borates
Trialkylboranes, BR3, can be oxidized to the corresponding borates, B(OR)3. One method for the determination of the amount of C-B bonds in a compound is by oxidation of R3B with trimethylamine oxide (Me3NO) to B(OR)3. The trimethylamine (Me3N) formed can then be titrated.
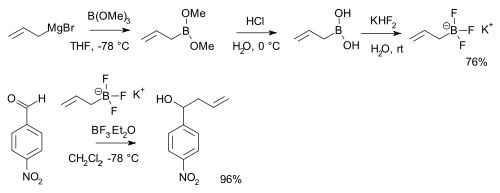
Boronic acids RB(OH)2 react with potassium bifluoride K[HF2] to form trifluoroborate salts K[RBF3][25] which are precursors to nucleophilic alkyl and aryl boron difluorides, ArBF2.[26] The salts are more stable than the boronic acids themselves and used for instance in alkylation of certain aldehydes:[27][note 1]
Suzuki reaction and related reactions
Organoboron compounds also lend themselves to transmetalation reactions, especially with organopalladium compounds. This reaction type is exemplified in the Suzuki reaction, which involves coupling of aryl- or vinyl-boronic acid with an aryl- or vinyl-halide catalyzed by a palladium(0) complex,[28]
-
(1)

This reaction is an important method for making carbon-carbon bonds.
Mechanism and stereochemistry
Boranes alone are generally not nucleophilic enough to transfer an alkyl group to an electrophilic center. However, after nucleophilic attack, the resulting borate is highly nucleophilic.[20] If the nucleophile contains unsaturated functionality or a leaving group at the α position, one of the R groups attached to boron is able to migrate to the electrophilic α carbon (see equation (2) below). The propensity of an organic group to migrate depends on its ability to stabilize negative charge: alkynyl > aryl ≈ alkenyl > primary alkyl > secondary alkyl > tertiary alkyl.[29] Migration takes place with retention of configuration at the migrating carbon[30] and inversion of configuration at the migration terminus (provided it is sp3 hybridized).[31] Bis(norbornyl)borane and 9-BBN are often used as "dummy" hydroboration reagents for this reason—only the R group derived from the hydroborated olefin is likely to migrate upon nucleophilic activation.
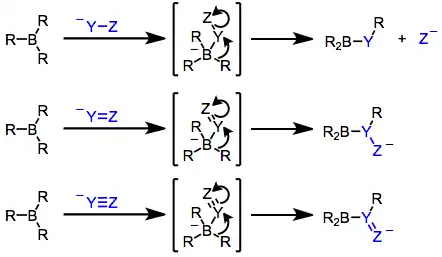
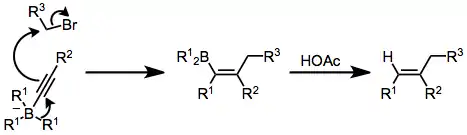
α-Halo enolates are commonly used as nucleophiles in this context. After nucleophilic attack at boron, the resulting ketoboronate rearranges to a neutral enolborane. Upon protonolysis, a functionalized carbonyl compound results.[32] The intermediate enolboranes may also be quenched with electrophiles.
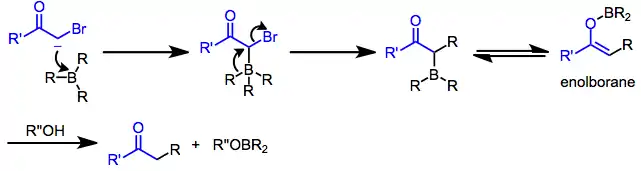
Alkynylboronates are versatile intermediates that can be converted to either ketones or olefins after simultaneous migration and attack of the alkyne on a separate electrophile. The electrophile and migrating group end up trans in the resulting alkenylborane. Protonolysis of this intermediate generates olefins,[33] while oxidation leads to ketones after tautomerization.[34]
Scope and limitations of reactions
The scope of organoboranes and borates as reagents for organic synthesis is extremely wide. Reactions of organoboron compounds may produce alcohols, carbonyl compounds, halides, peroxides, amines, and other functionality depending on other starting materials employed and reaction conditions. This section covers a small subset of these methods, focusing on the synthesis of alcohols, carbonyl compounds, and halides.
Alcohol synthesis from organoboranes and borates relies on either nucleophilic group transfer to a carbonyl group or oxidation of an intermediate organoborane. Homologated primary alcohols result from the treatment of organoboranes with carbon monoxide and a hydride.[35]
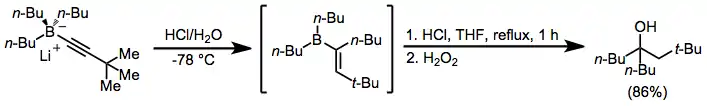
Tertiary alcohols with two identical groups attached to the alcohol carbon may be synthesized through a double migration reaction of alkynylborates in the presence of acid.[34] Use of a single equivalent of acid and oxidation or protonolysis leads to ketones or olefins, respectively (see Mechanism and Stereochemistry section above).
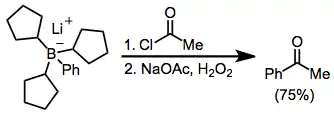
Acylation of borates is possible in the presence of an acyl halide. Here, the borate was generated from tri(cyclopentyl)borane and phenyllithium; the three cyclopentyl groups are serving as "dummy" groups and do not migrate to a significant amount.[36]
Treatment of trialkylboranes with α-halo enolates leads to functionalized ketones.[32] Because the migration is stereospecific (retentive with respect to the migrating group and invertive at the α carbon), this method provides a means for the synthesis of enantiopure α-alkyl or -aryl ketones.[37]
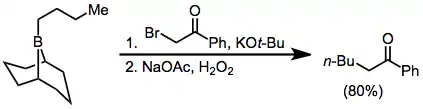
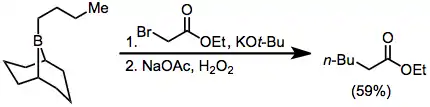
α-Halo ester enolates also add to boranes to eventually afford α-functionalized products; however, yields are slightly lower.[38] Diazoesters and diazoketones may also be used in this context without the requirement for external base.[39] α,α'-Dialo enolates react with boranes to form α-halo carbonyl compounds that can be further functionalized at the α position.[40]
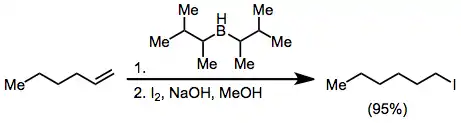
Halides may be synthesized from organoboranes by activating with hydroxide or alkoxide and treatment with X2. Two of the three alkyl groups attached to the borane may be converted to halide in the presence of excess base, but the use of disiamylborane as the hydroborating reagent permits the selective halogenation of only the hydroborated olefin.[41]
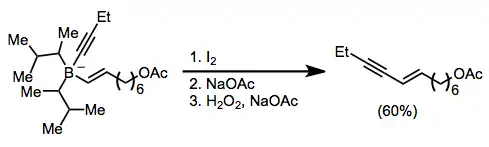
Treatment of an alkenylborane with iodine or bromine leads to migration of one of the organic groups attached to boron. Alkynyl groups migrate selectively, forming enynes after treatment with sodium acetate and hydrogen peroxide.[42]
Other uses
TEB – Triethylborane was used to ignite the JP-7 fuel of the Pratt & Whitney J58 variable cycle engines powering the Lockheed SR-71 Blackbird.
Notes
- Displayed is a reaction sequence starting with reaction of allyl magnesium bromide with trimethyl borate, followed by hydrolysis of the boronic ester to the boronic acid with hydrochloric acid. The aldehyde is p-nitrobenzaldehyde.
References
- The Roles of Boron and Silicon, Susan E. Thomas; Oxford Chemistry Primers No.1; 1991: Very good general book covering all the important reactions of boron and organoboranes in organic chemistry.
- Organometallics Christoph Elschenbroich 3rd Ed. 2006 ISBN 3-527-29390-6 – Wiley-VCH, Weinheim
- Negishi, E.-i.; Idacavage, M. J. Org. React. 1985, 33, 1. doi:10.1002/0471264180.or033.01
- Advanced Organic Chemistry, F.A. carey, R.J. Sundberg ISBN 0-306-41088-5
- Brown, H. C. “Organic Syntheses via Boranes” John Wiley & Sons, Inc. New York: 1975. ISBN 0-471-11280-1.
- Bartlett, Ruth A.; Dias, H. V. Rasika; Olmstead, Marilyn M.; Power, Philip P.; Weese, Kenneth J. (1990). "Synthesis of the monomeric HBtrip2 (Trip - 2,4,6-iso-Pr3C6H2) and the x-ray crystal structures of [HBMes2]2 (Mes = 2,4,6,-Me3C6H2) and HBtrip2". Organometallics. 9: 146–150. doi:10.1021/om00115a023.
- Grimes, R. N. (2016). Carboranes, 3rd edition. New York: Academic Press. ISBN 9780128019054.
- Segawa Yasutomo, Yamashita Makoto, Nozaki Kyoko (2006). "Boryllithium: Isolation, Characterization, and Reactivity as a Boryl Anion". Science. 314 (5796): 113–115. Bibcode:2006Sci...314..113S. doi:10.1126/science.1131914. PMID 17023656.CS1 maint: multiple names: authors list (link)
- Bethany Halford Boron Attacks Electropositive element pressed into action as nucleophilic boryllithium Chemical & Engineering News 2006; Volume 84(41): 11 Link
- Boronic Acids: Preparation, Applications in Organic Synthesis and Medicine. Dennis G. Hall ISBN 3-527-30991-8
- Paetzold Peter, Englert Ulli, Finger Rudolf, Schmitz Thomas, Tapper Alexander, Ziembinski Ralf (2004). "Reactions at the Boron-Carbon Double Bond of Methyl(methylidene)boranes". Z. Anorg. Allg. Chem. 630 (4): 508–518. doi:10.1002/zaac.200300396.CS1 maint: multiple names: authors list (link)
- Curran D. P., Solovyev A., Makhlouf Brahmi M., Fensterbank L., Malacria M., Lacôte E. (2011). "Synthesis and Reactions of N-Heterocyclic Carbene Boranes". Angewandte Chemie International Edition. 50 (44): 10294–10317. doi:10.1002/anie.201102717. PMID 21898724.CS1 maint: multiple names: authors list (link)
- Yuzhong Wang, Brandon Quillian, Pingrong Wei, Chaitanya S. Wannere, Yaoming Xie, R. Bruce King, Henry F. Schaefer, III, Paul v. R. Schleyer, and Gregory H. Robinson (2007). "A Stable Neutral Diborene Containing a B=B Double Bond". J. Am. Chem. Soc. 129 (41): 12412–12413. doi:10.1021/ja075932i. PMID 17887683.CS1 maint: uses authors parameter (link)
- Neutral Diborene Is A First Ron Dagani Chemical & Engineering News October 1, 2007 Volume 85, Number 40 p. 10
- The boron precursor is boron tribromide and the reducing agent is KC8 which abstracts the required protons from diethyl ether solvent.
- Braunschweig, Holger; Dewhurst, Rian D. (2013-03-25). "Single, Double, Triple Bonds and Chains: The Formation of Electron-Precise B-B Bonds". Angewandte Chemie International Edition. 52 (13): 3574–3583. doi:10.1002/anie.201208189. ISSN 1521-3773. PMID 23362015.
- Arrowsmith, Merle; Braunschweig, Holger; Stennett, Tom E. (2017-01-02). "Formation and Reactivity of Electron-Precise B−B Single and Multiple Bonds". Angewandte Chemie International Edition. 56 (1): 96–115. doi:10.1002/anie.201610072. ISSN 1521-3773. PMID 27860056.
- Allred, A. L.; Rochow, E. G. J. Inorg. Nucl. Chem. 1958, 5, 264.
- Negishi, E.-i.; Idacavage, M. J. Org. React. 1985, 33, 1. doi:10.1002/0471264180.or033.01
- Negishi, E.-i. J. Organometal. Chem. 1976, 108, 281.
- Jacob, III, P.; Brown, H. C. J. Org. Chem. 1977, 42, 579.
- Rathke, M. W.; Brown, H. C. J. Am. Chem. Soc. 1967, 89, 2740.
- Lachance H., Hall D. (2008). Allylboration of Carbonyl Compounds. Org. React. 73. p. 1. doi:10.1002/0471264180.or073.01. ISBN 978-0471264187.
- Nicolaou, K.C.; Sarabia, F.; Ninkovic, S.; Finlay, M.R.V.; Boddy, C.N.C. (1998). "Probing the Ring Size of Epothilones: Total Synthesis of 14-, 15-, 17-, and 18 Epothilones A". Angewandte Chemie International Edition in English. 37 (1–2): 81–84. doi:10.1002/(sici)1521-3773(19980202)37:1/2<81::aid-anie81>3.0.co;2-c. Retrieved 2008-03-02.
- Vedejs E., Chapman R. W., Fields S. C., Lin S., Schrimpf M. R. (1995). "Conversion of Arylboronic Acids into Potassium Aryltrifluoroborates: Convenient Precursors of Arylboron Difluoride Lewis Acids". J. Org. Chem. 60 (10): 3020–3027. doi:10.1021/jo00115a016.CS1 maint: multiple names: authors list (link)
- Molander Gary A., Canturk Belgin (2009). "Organotrifluoroborates and Monocoordinated Palladium Complexes as Catalysts—A Perfect Combination for Suzuki–Miyaura Coupling". Angew. Chem. Int. Ed. 48 (49): 9240–9261. doi:10.1002/anie.200904306. PMC 2917751. PMID 19899086.
- Batey Robert A., Quach Tan D., Shen Ming, Thadani Avinash N., Smil David V., Li Sze-Wan, MacKay D. Bruce (2002). "Organoboron compounds as mild nucleophiles in Lewis acid- and transition metal-catalyzed C–C bond-forming reactions" (PDF). Pure Appl. Chem. 74 (1): 43–55. doi:10.1351/pac200274010043.CS1 maint: multiple names: authors list (link)
- Miyaura, Norio; Suzuki, Akira (1995). "Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds". Chemical Reviews. 95 (7): 2457–2483. CiteSeerX 10.1.1.735.7660. doi:10.1021/cr00039a007.
- Miyaura, M.; Sasaki, N.; Itoh, M.; Suzuki, A. Tetrahedron Lett. 1977, 173.
- Zweifel, G. in Aspects of Mechanism and Organometallic Chemistry, J. H. Bewster, Ed., Plenum, 1978, p. 229.
- Midland, M. M.; Zolopa, A. R.; Halterman, R. L. J. Am. Chem. Soc. 1979, 101, 248.
- Brown, H. C.; Rogi, M. M.; Nambu, H.; Rathke, M. W. J. Am. Chem. Soc. 1969, 91, 2147.
- Corey, E. J.; Ravindranathan, T. J. Am. Chem. Soc. 1972, 94, 4013.
- Midland, M. M.; Brown, H. C. J. Org. Chem. 1975, 40, 2845.
- Rathke, M. W.; Brown, H. C. J. Am. Chem. Soc. 1967, 89, 2740.
- Negishi, E.-i.; Abramovitch, A.; Merrill, R. E. Chem. Commun. 1975, 138.
- Nesmeyanov, A. N.; Sokolik, R. A. The Organic Compounds of Boron, Aluminium, Gallium, Indium, and Thallium, North-Holland, Amsterdam, 1967.
- Brown, H. C.; Rogic, M. M.; Rathke, M. W.; Kabalka, G. W. J. Am. Chem. Soc. 1968, 90, 818.
- Hooz, J.; Gunn, D. M. J. Am. Chem. Soc. 1969, 91, 6195.
- Pasto, D. J.; Wojtkowski, P. W. J. Org. Chem. 1971, 36, 1790.
- Brown, H. C.; Rathke, M. W.; Rogic, M. M. J. Am. Chem. Soc. 1968, 90, 5038.
- Negishi, E.-i.; Lew, G.; Yoshida, T. Chem. Commun. 1973, 874.
Compounds of carbon with other elements in the periodic table | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Legend |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||