Fluorine
Fluorine is a chemical element with the symbol F and atomic number 9. It is the lightest halogen and exists as a highly toxic pale yellow diatomic gas at standard conditions. As the most electronegative element, it is extremely reactive, as it reacts with all other elements, except for argon, neon, and helium.
 Liquid fluorine (at extremely low temperatures) | ||||||||||||||||||
| Fluorine | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pronunciation |
| |||||||||||||||||
| Allotropes | alpha, beta | |||||||||||||||||
| Appearance | gas: very pale yellow liquid: bright yellow solid: alpha is opaque, beta is transparent | |||||||||||||||||
| Standard atomic weight Ar, std(F) | 18.998403163(6)[1] | |||||||||||||||||
| Fluorine in the periodic table | ||||||||||||||||||
| ||||||||||||||||||
| Atomic number (Z) | 9 | |||||||||||||||||
| Group | group 17 (halogens) | |||||||||||||||||
| Period | period 2 | |||||||||||||||||
| Block | p-block | |||||||||||||||||
| Electron configuration | [He] 2s2 2p5[2] | |||||||||||||||||
| Electrons per shell | 2, 7 | |||||||||||||||||
| Physical properties | ||||||||||||||||||
| Phase at STP | gas | |||||||||||||||||
| Melting point | (F2) 53.48 K (−219.67 °C, −363.41 °F)[3] | |||||||||||||||||
| Boiling point | (F2) 85.03 K (−188.11 °C, −306.60 °F)[3] | |||||||||||||||||
| Density (at STP) | 1.696 g/L[4] | |||||||||||||||||
| when liquid (at b.p.) | 1.505 g/cm3[5] | |||||||||||||||||
| Triple point | 53.48 K, 90 kPa[3] | |||||||||||||||||
| Critical point | 144.41 K, 5.1724 MPa[3] | |||||||||||||||||
| Heat of vaporization | 6.51 kJ/mol[4] | |||||||||||||||||
| Molar heat capacity | Cp: 31 J/(mol·K)[5] (at 21.1 °C) Cv: 23 J/(mol·K)[5] (at 21.1 °C) | |||||||||||||||||
Vapor pressure
| ||||||||||||||||||
| Atomic properties | ||||||||||||||||||
| Oxidation states | −1 (oxidizes oxygen) | |||||||||||||||||
| Electronegativity | Pauling scale: 3.98[2] | |||||||||||||||||
| Ionization energies | ||||||||||||||||||
| Covalent radius | 64 pm[7] | |||||||||||||||||
| Van der Waals radius | 135 pm[8] | |||||||||||||||||
Color lines in a spectral range | ||||||||||||||||||
| Other properties | ||||||||||||||||||
| Natural occurrence | primordial | |||||||||||||||||
| Crystal structure | cubic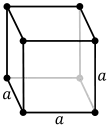 | |||||||||||||||||
| Thermal conductivity | 0.02591 W/(m·K)[9] | |||||||||||||||||
| Magnetic ordering | diamagnetic (−1.2×10−4)[10][11] | |||||||||||||||||
| CAS Number | 7782-41-4[2] | |||||||||||||||||
| History | ||||||||||||||||||
| Naming | after the mineral fluorite, itself named after Latin fluo (to flow, in smelting) | |||||||||||||||||
| Discovery | André-Marie Ampère (1810) | |||||||||||||||||
| First isolation | Henri Moissan[2] (June 26, 1886) | |||||||||||||||||
| Named by | Humphry Davy | |||||||||||||||||
| Main isotopes of fluorine[12] | ||||||||||||||||||
| ||||||||||||||||||
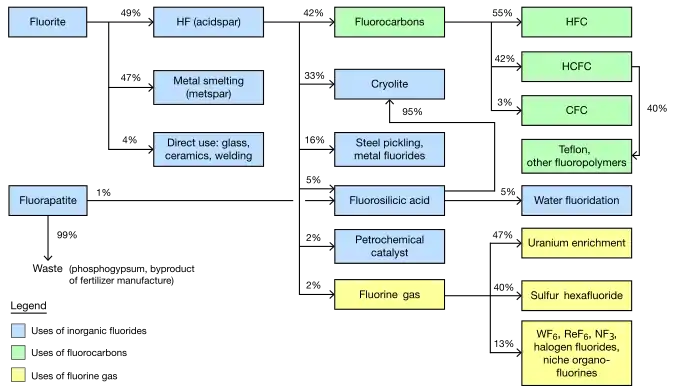
Among the elements, fluorine ranks 24th in universal abundance and 13th in terrestrial abundance. Fluorite, the primary mineral source of fluorine which gave the element its name, was first described in 1529; as it was added to metal ores to lower their melting points for smelting, the Latin verb fluo meaning "flow" gave the mineral its name. Proposed as an element in 1810, fluorine proved difficult and dangerous to separate from its compounds, and several early experimenters died or sustained injuries from their attempts. Only in 1886 did French chemist Henri Moissan isolate elemental fluorine using low-temperature electrolysis, a process still employed for modern production. Industrial production of fluorine gas for uranium enrichment, its largest application, began during the Manhattan Project in World War II.
Owing to the expense of refining pure fluorine, most commercial applications use fluorine compounds, with about half of mined fluorite used in steelmaking. The rest of the fluorite is converted into corrosive hydrogen fluoride en route to various organic fluorides, or into cryolite, which plays a key role in aluminium refining. Molecules containing a carbon–fluorine bond often have very high chemical and thermal stability; their major uses are as refrigerants, electrical insulation and cookware, the last as PTFE (Teflon). Pharmaceuticals such as atorvastatin and fluoxetine contain C−F bonds. The fluoride ion from dissolved fluoride salts inhibits dental cavities, and so finds use in toothpaste and water fluoridation. Global fluorochemical sales amount to more than US$15 billion a year.
Fluorocarbon gases are generally greenhouse gases with global-warming potentials 100 to 23,500 times that of carbon dioxide, SF6 having the highest global warming potential of any known substance. Organofluorine compounds often persist in the environment due to the strength of the carbon–fluorine bond. Fluorine has no known metabolic role in mammals; a few plants and sea sponges synthesize organofluorine poisons (most often monofluoroacetates) that help deter predation.[13]
Characteristics
Electron configuration
Fluorine atoms have nine electrons, one fewer than neon, and electron configuration 1s22s22p5: two electrons in a filled inner shell and seven in an outer shell requiring one more to be filled. The outer electrons are ineffective at nuclear shielding, and experience a high effective nuclear charge of 9 − 2 = 7; this affects the atom's physical properties.[2]
Fluorine's first ionization energy is third-highest among all elements, behind helium and neon,[14] which complicates the removal of electrons from neutral fluorine atoms. It also has a high electron affinity, second only to chlorine,[15] and tends to capture an electron to become isoelectronic with the noble gas neon;[2] it has the highest electronegativity of any element.[16] Fluorine atoms have a small covalent radius of around 60 picometers, similar to those of its period neighbors oxygen and neon.[17][18][note 1]
Reactivity
| External video | |
|---|---|
.png.webp)
The bond energy of difluorine is much lower than that of either Cl
2 or Br
2 and similar to the easily cleaved peroxide bond; this, along with high electronegativity, accounts for fluorine's easy dissociation, high reactivity, and strong bonds to non-fluorine atoms.[19][20] Conversely, bonds to other atoms are very strong because of fluorine's high electronegativity. Unreactive substances like powdered steel, glass fragments, and asbestos fibers react quickly with cold fluorine gas; wood and water spontaneously combust under a fluorine jet.[4][21]
Reactions of elemental fluorine with metals require varying conditions. Alkali metals cause explosions and alkaline earth metals display vigorous activity in bulk; to prevent passivation from the formation of metal fluoride layers, most other metals such as aluminium and iron must be powdered,[19] and noble metals require pure fluorine gas at 300–450 °C (575–850 °F).[22] Some solid nonmetals (sulfur, phosphorus) react vigorously in liquid fluorine.[23] Hydrogen sulfide[23] and sulfur dioxide[24] combine readily with fluorine, the latter sometimes explosively; sulfuric acid exhibits much less activity, requiring elevated temperatures.[25]
Hydrogen, like some of the alkali metals, reacts explosively with fluorine.[26] Carbon, as lamp black, reacts at room temperature to yield fluoromethane. Graphite combines with fluorine above 400 °C (750 °F) to produce non-stoichiometric carbon monofluoride; higher temperatures generate gaseous fluorocarbons, sometimes with explosions.[27] Carbon dioxide and carbon monoxide react at or just above room temperature,[28] whereas paraffins and other organic chemicals generate strong reactions:[29] even completely substituted haloalkanes such as carbon tetrachloride, normally incombustible, may explode. Although nitrogen trifluoride is stable, nitrogen requires an electric discharge at elevated temperatures for reaction with fluorine to occur, due to the very strong triple bond in elemental nitrogen;[31] ammonia may react explosively.[32][33] Oxygen does not combine with fluorine under ambient conditions, but can be made to react using electric discharge at low temperatures and pressures; the products tend to disintegrate into their constituent elements when heated.[34][35][36] Heavier halogens[37] react readily with fluorine as does the noble gas radon;[38] of the other noble gases, only xenon and krypton react, and only under special conditions.[39]
Phases
2 molecules that may assume any angle. Other molecules are constrained to planes.
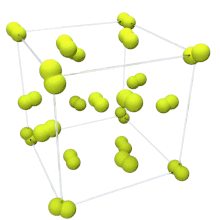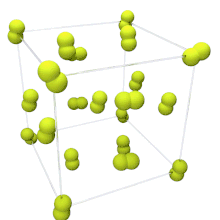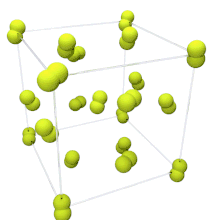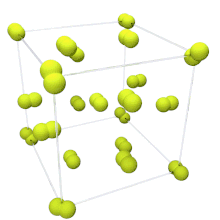
At room temperature, fluorine is a gas of diatomic molecules,[4] pale yellow when pure (sometimes described as yellow-green).[40] It has a characteristic halogen-like pungent and biting odor detectable at 20 ppb.[41] Fluorine condenses into a bright yellow liquid at −188 °C (−306 °F), a transition temperature similar to those of oxygen and nitrogen.[42]
Fluorine has two solid forms, α- and β-fluorine. The latter crystallizes at −220 °C (−364 °F) and is transparent and soft, with the same disordered cubic structure of freshly crystallized solid oxygen,[42][note 2] unlike the orthorhombic systems of other solid halogens.[46][47] Further cooling to −228 °C (−378 °F) induces a phase transition into opaque and hard α-fluorine, which has a monoclinic structure with dense, angled layers of molecules. The transition from β- to α-fluorine is more exothermic than the condensation of fluorine, and can be violent.[46][47][note 3]
Isotopes
Only one isotope of fluorine occurs naturally in abundance, the stable isotope 19
F.[48] It has a high magnetogyric ratio[note 4] and exceptional sensitivity to magnetic fields; because it is also the only stable isotope, it is used in magnetic resonance imaging.[50] Seventeen radioisotopes with mass numbers from 14 to 31 have been synthesized, of which 18
F is the most stable with a half-life of 109.77 minutes. Other radioisotopes have half-lives less than 70 seconds; most decay in less than half a second.[51] The isotopes 17
F and 18
F undergo β+ decay and electron capture, lighter isotopes decay by proton emission, and those heavier than 19
F undergo β− decay (the heaviest ones with delayed neutron emission).[51][52] Two metastable isomers of fluorine are known, 18m
F, with a half-life of 162(7) nanoseconds, and 26m
F, with a half-life of 2.2(1) milliseconds.[53]
Occurrence
Universe
| Atomic number |
Element | Relative amount |
|---|---|---|
| 6 | Carbon | 4,800 |
| 7 | Nitrogen | 1,500 |
| 8 | Oxygen | 8,800 |
| 9 | Fluorine | 1 |
| 10 | Neon | 1,400 |
| 11 | Sodium | 24 |
| 12 | Magnesium | 430 |
Among the lighter elements, fluorine's abundance value of 400 ppb (parts per billion) – 24th among elements in the universe – is exceptionally low: other elements from carbon to magnesium are twenty or more times as common.[55] This is because stellar nucleosynthesis processes bypass fluorine, and any fluorine atoms otherwise created have high nuclear cross sections, allowing further fusion with hydrogen or helium to generate oxygen or neon respectively.[55][56]
Beyond this transient existence, three explanations have been proposed for the presence of fluorine:[55][57]
- during type II supernovae, bombardment of neon atoms by neutrinos could transmute them to fluorine;
- the solar wind of Wolf–Rayet stars could blow fluorine away from any hydrogen or helium atoms; or
- fluorine is borne out on convection currents arising from fusion in asymptotic giant branch stars.
Earth
Fluorine is the thirteenth most common element in Earth's crust at 600–700 ppm (parts per million) by mass.[58] Elemental fluorine does not occur naturally.[59][60] Instead, all fluorine exists as fluoride-containing minerals. Fluorite, fluorapatite and cryolite are the most industrially significant.[58][61] Fluorite, also known as fluorspar, (CaF
2), abundant worldwide, is the main source of fluoride, and hence fluorine. China and Mexico are the major suppliers.[61][62][63][64][65] Fluorapatite (Ca5(PO4)3F), which contains most of the world's fluoride, is an inadvertent source of fluoride as a byproduct of fertilizer production.[61] Cryolite (Na
3AlF
6), used in the production of aluminium, is the most fluorine-rich mineral. Economically viable natural sources of cryolite have been exhausted, and most is now synthesised commercially.[61]
 Fluorite: Pink globular mass with crystal facets
Fluorite: Pink globular mass with crystal facets Fluorapatite: Long prism-like crystal, without luster, at an angle coming out of aggregate-like rock
Fluorapatite: Long prism-like crystal, without luster, at an angle coming out of aggregate-like rock Cryolite: A parallelogram-shaped outline with diatomic molecules arranged in two layers
Cryolite: A parallelogram-shaped outline with diatomic molecules arranged in two layers
Other minerals such as topaz contain fluorine. Fluorides, unlike other halides, are insoluble and do not occur in commercially favorable concentrations in saline waters.[61] Trace quantities of organofluorines of uncertain origin have been detected in volcanic eruptions and geothermal springs.[66] The existence of gaseous fluorine in crystals, suggested by the smell of crushed antozonite, is contentious;[67][68] a 2012 study reported the presence of 0.04% F
2 by weight in antozonite, attributing these inclusions to radiation from the presence of tiny amounts of uranium.[68]
History

Early discoveries
In 1529, Georgius Agricola described fluorite as an additive used to lower the melting point of metals during smelting.[69][70][note 5] He penned the Latin word fluorēs (fluor, flow) for fluorite rocks. The name later evolved into fluorspar (still commonly used) and then fluorite.[62][74][75] The composition of fluorite was later determined to be calcium difluoride.[76]
Hydrofluoric acid was used in glass etching from 1720 onwards.[note 6] Andreas Sigismund Marggraf first characterized it in 1764 when he heated fluorite with sulfuric acid, and the resulting solution corroded its glass container.[78][79] Swedish chemist Carl Wilhelm Scheele repeated the experiment in 1771, and named the acidic product fluss-spats-syran (fluorspar acid).[79][80] In 1810, the French physicist André-Marie Ampère suggested that hydrogen and an element analogous to chlorine constituted hydrofluoric acid.[81] He also proposed in a letter to Sir Humphry Davy dated August 26, 1812 that this then-unknown substance may be named fluorine from fluoric acid and the -ine suffix of other halogens.[82][83] This word, often with modifications, is used in most European languages; however, Greek, Russian, and some others, following Ampère's later suggestion, use the name ftor or derivatives, from the Greek φθόριος (phthorios, destructive).[84] The New Latin name fluorum gave the element its current symbol F; Fl was used in early papers.[85][note 7]
Isolation
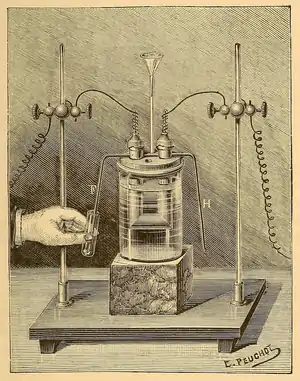
Initial studies on fluorine were so dangerous that several 19th-century experimenters were deemed "fluorine martyrs" after misfortunes with hydrofluoric acid.[note 8] Isolation of elemental fluorine was hindered by the extreme corrosiveness of both elemental fluorine itself and hydrogen fluoride, as well as the lack of a simple and suitable electrolyte.[76][86] Edmond Frémy postulated that electrolysis of pure hydrogen fluoride to generate fluorine was feasible and devised a method to produce anhydrous samples from acidified potassium bifluoride; instead, he discovered that the resulting (dry) hydrogen fluoride did not conduct electricity.[76][86][87] Frémy's former student Henri Moissan persevered, and after much trial and error found that a mixture of potassium bifluoride and dry hydrogen fluoride was a conductor, enabling electrolysis. To prevent rapid corrosion of the platinum in his electrochemical cells, he cooled the reaction to extremely low temperatures in a special bath and forged cells from a more resistant mixture of platinum and iridium, and used fluorite stoppers.[86][88] In 1886, after 74 years of effort by many chemists, Moissan isolated elemental fluorine.[87][89]
In 1906, two months before his death, Moissan received the Nobel Prize in Chemistry,[90] with the following citation:[86]
[I]n recognition of the great services rendered by him in his investigation and isolation of the element fluorine ... The whole world has admired the great experimental skill with which you have studied that savage beast among the elements.[note 9]
Later uses

The Frigidaire division of General Motors (GM) experimented with chlorofluorocarbon refrigerants in the late 1920s, and Kinetic Chemicals was formed as a joint venture between GM and DuPont in 1930 hoping to market Freon-12 (CCl
2F
2) as one such refrigerant. It replaced earlier and more toxic compounds, increased demand for kitchen refrigerators, and became profitable; by 1949 DuPont had bought out Kinetic and marketed several other Freon compounds.[79][91][92][93] Polytetrafluoroethylene (Teflon) was serendipitously discovered in 1938 by Roy J. Plunkett while working on refrigerants at Kinetic, and its superlative chemical and thermal resistance lent it to accelerated commercialization and mass production by 1941.[79][91][92]
Large-scale production of elemental fluorine began during World War II. Germany used high-temperature electrolysis to make tons of the planned incendiary chlorine trifluoride[94] and the Manhattan Project used huge quantities to produce uranium hexafluoride for uranium enrichment. Since UF
6 is as corrosive as fluorine, gaseous diffusion plants required special materials: nickel for membranes, fluoropolymers for seals, and liquid fluorocarbons as coolants and lubricants. This burgeoning nuclear industry later drove post-war fluorochemical development.[95]
Compounds
Fluorine has a rich chemistry, encompassing organic and inorganic domains. It combines with metals, nonmetals, metalloids, and most noble gases,[96] and almost exclusively assumes an oxidation state of −1.[note 10] Fluorine's high electron affinity results in a preference for ionic bonding; when it forms covalent bonds, these are polar, and almost always single.[99][100][note 11]
Metals
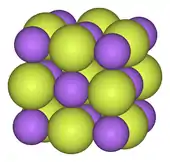
Alkali metals form ionic and highly soluble monofluorides; these have the cubic arrangement of sodium chloride and analogous chlorides.[101][102] Alkaline earth difluorides possess strong ionic bonds but are insoluble in water,[85] with the exception of beryllium difluoride, which also exhibits some covalent character and has a quartz-like structure.[103] Rare earth elements and many other metals form mostly ionic trifluorides.[104][105][106]
Covalent bonding first comes to prominence in the tetrafluorides: those of zirconium, hafnium[107][108] and several actinides[109] are ionic with high melting points,[110][note 12] while those of titanium,[113] vanadium,[114] and niobium are polymeric,[115] melting or decomposing at no more than 350 °C (660 °F).[116] Pentafluorides continue this trend with their linear polymers and oligomeric complexes.[117][118][119] Thirteen metal hexafluorides are known,[note 13] all octahedral, and are mostly volatile solids but for liquid MoF
6 and ReF
6, and gaseous WF
6.[120][121][122] Rhenium heptafluoride, the only characterized metal heptafluoride, is a low-melting molecular solid with pentagonal bipyramidal molecular geometry.[123] Metal fluorides with more fluorine atoms are particularly reactive.[124]
| Structural progression of metal fluorides | ||
 |
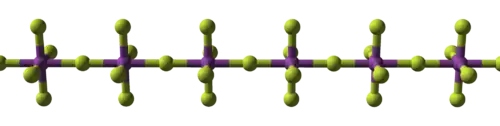 |
 |
| Sodium fluoride, ionic | Bismuth pentafluoride, polymeric | Rhenium heptafluoride, molecular |
Hydrogen
Hydrogen and fluorine combine to yield hydrogen fluoride, in which discrete molecules form clusters by hydrogen bonding, resembling water more than hydrogen chloride.[125][126][127] It boils at a much higher temperature than heavier hydrogen halides and unlike them is miscible with water.[128] Hydrogen fluoride readily hydrates on contact with water to form aqueous hydrogen fluoride, also known as hydrofluoric acid. Unlike the other hydrohalic acids, which are strong, hydrofluoric acid is a weak acid at low concentrations.[129][note 14] However, it can attack glass, something the other acids cannot do.[131]
Other reactive nonmetals

Binary fluorides of metalloids and p-block nonmetals are generally covalent and volatile, with varying reactivities. Period 3 and heavier nonmetals can form hypervalent fluorides.[133]
Boron trifluoride is planar and possesses an incomplete octet. It functions as a Lewis acid and combines with Lewis bases like ammonia to form adducts.[134] Carbon tetrafluoride is tetrahedral and inert;[note 15] its group analogues, silicon and germanium tetrafluoride, are also tetrahedral[135] but behave as Lewis acids.[136][137] The pnictogens form trifluorides that increase in reactivity and basicity with higher molecular weight, although nitrogen trifluoride resists hydrolysis and is not basic.[138] The pentafluorides of phosphorus, arsenic, and antimony are more reactive than their respective trifluorides, with antimony pentafluoride the strongest neutral Lewis acid known.[117][139][140]
Chalcogens have diverse fluorides: unstable difluorides have been reported for oxygen (the only known compound with oxygen in an oxidation state of +2), sulfur, and selenium; tetrafluorides and hexafluorides exist for sulfur, selenium, and tellurium. The latter are stabilized by more fluorine atoms and lighter central atoms, so sulfur hexafluoride is especially inert.[141][142] Chlorine, bromine, and iodine can each form mono-, tri-, and pentafluorides, but only iodine heptafluoride has been characterized among possible interhalogen heptafluorides.[143] Many of them are powerful sources of fluorine atoms, and industrial applications using chlorine trifluoride require precautions similar to those using fluorine.[144][145]
Noble gases

Noble gases, having complete electron shells, defied reaction with other elements until 1962 when Neil Bartlett reported synthesis of xenon hexafluoroplatinate;[147] xenon difluoride, tetrafluoride, hexafluoride, and multiple oxyfluorides have been isolated since then.[148] Among other noble gases, krypton forms a difluoride,[149] and radon and fluorine generate a solid suspected to be radon difluoride.[150][151] Binary fluorides of lighter noble gases are exceptionally unstable: argon and hydrogen fluoride combine under extreme conditions to give argon fluorohydride.[39] Helium and neon have no long-lived fluorides,[152] and no neon fluoride has ever been observed;[153] helium fluorohydride has been detected for milliseconds at high pressures and low temperatures.[152]
Organic compounds

The carbon–fluorine bond is organic chemistry's strongest,[155] and gives stability to organofluorines.[156] It is almost non-existent in nature, but is used in artificial compounds. Research in this area is usually driven by commercial applications;[157] the compounds involved are diverse and reflect the complexity inherent in organic chemistry.[91]
Discrete molecules
The substitution of hydrogen atoms in an alkane by progressively more fluorine atoms gradually alters several properties: melting and boiling points are lowered, density increases, solubility in hydrocarbons decreases and overall stability increases. Perfluorocarbons,[note 16] in which all hydrogen atoms are substituted, are insoluble in most organic solvents, reacting at ambient conditions only with sodium in liquid ammonia.[158]
The term perfluorinated compound is used for what would otherwise be a perfluorocarbon if not for the presence of a functional group,[159][note 17] often a carboxylic acid. These compounds share many properties with perfluorocarbons such as stability and hydrophobicity,[161] while the functional group augments their reactivity, enabling them to adhere to surfaces or act as surfactants;[162] Fluorosurfactants, in particular, can lower the surface tension of water more than their hydrocarbon-based analogues. Fluorotelomers, which have some unfluorinated carbon atoms near the functional group, are also regarded as perfluorinated.[161]
Polymers
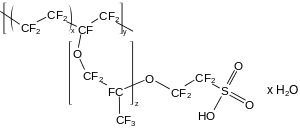
Polymers exhibit the same stability increases afforded by fluorine substitution (for hydrogen) in discrete molecules; their melting points generally increase too.[163] Polytetrafluoroethylene (PTFE), the simplest fluoropolymer and perfluoro analogue of polyethylene with structural unit –CF
2–, demonstrates this change as expected, but its very high melting point makes it difficult to mold.[164] Various PTFE derivatives are less temperature-tolerant but easier to mold: fluorinated ethylene propylene replaces some fluorine atoms with trifluoromethyl groups, perfluoroalkoxy alkanes do the same with trifluoromethoxy groups,[164] and Nafion contains perfluoroether side chains capped with sulfonic acid groups.[165][166] Other fluoropolymers retain some hydrogen atoms; polyvinylidene fluoride has half the fluorine atoms of PTFE and polyvinyl fluoride has a quarter, but both behave much like perfluorinated polymers.[167]
Production
Elemental fluorine and virtually all fluorine compounds are produced from hydrogen fluoride or its aqueous solutions, hydrofluoric acid. Hydrogen fluoride is produced in kilns by the endothermic reaction of fluorite (CaF2) with sulfuric acid:[168]
- CaF2 + H2SO4 → 2 HF(g) + CaSO4
The gaseous HF can then be absorbed in water or liquefied.[169]
About 20% of manufactured HF is a byproduct of fertilizer production, which produces hexafluorosilicic acid (H2SiF6), which can be degraded to release HF thermally and by hydrolysis:
- H2SiF6 → 2 HF + SiF4
- SiF4 + 2 H2O → 4 HF + SiO2
Industrial routes to F2

Moissan's method is used to produce industrial quantities of fluorine, via the electrolysis of a potassium fluoride/hydrogen fluoride mixture: hydrogen and fluoride ions are reduced and oxidized at a steel container cathode and a carbon block anode, under 8–12 volts, to generate hydrogen and fluorine gas respectively.[63][170] Temperatures are elevated, KF•2HF melting at 70 °C (158 °F) and being electrolyzed at 70–130 °C (158–266 °F). KF, which acts as catalyst, is essential since pure HF cannot be electrolyzed.[79][171][172] Fluorine can be stored in steel cylinders that have passivated interiors, at temperatures below 200 °C (392 °F); otherwise nickel can be used.[79][173] Regulator valves and pipework are made of nickel, the latter possibly using Monel instead.[174] Frequent passivation, along with the strict exclusion of water and greases, must be undertaken. In the laboratory, glassware may carry fluorine gas under low pressure and anhydrous conditions;[174] some sources instead recommend nickel-Monel-PTFE systems.[175]
Laboratory routes
While preparing for a 1986 conference to celebrate the centennial of Moissan's achievement, Karl O. Christe reasoned that chemical fluorine generation should be feasible since some metal fluoride anions have no stable neutral counterparts; their acidification potentially triggers oxidation instead. He devised a method which evolves fluorine at high yield and atmospheric pressure:[176]
- 2 KMnO4 + 2 KF + 10 HF + 3 H2O2 → 2 K2MnF6 + 8 H2O + 3 O2↑
- 2 K2MnF6 + 4 SbF5 → 4 KSbF6 + 2 MnF3 + F2↑
Christe later commented that the reactants "had been known for more than 100 years and even Moissan could have come up with this scheme."[177] As late as 2008, some references still asserted that fluorine was too reactive for any chemical isolation.[178]
Industrial applications
Fluorite mining, which supplies most global fluorine, peaked in 1989 when 5.6 million metric tons of ore were extracted. Chlorofluorocarbon restrictions lowered this to 3.6 million tons in 1994; production has since been increasing. Around 4.5 million tons of ore and revenue of US$550 million were generated in 2003; later reports estimated 2011 global fluorochemical sales at $15 billion and predicted 2016–18 production figures of 3.5 to 5.9 million tons, and revenue of at least $20 billion.[79][179][180][181][182] Froth flotation separates mined fluorite into two main metallurgical grades of equal proportion: 60–85% pure metspar is almost all used in iron smelting whereas 97%+ pure acidspar is mainly converted to the key industrial intermediate hydrogen fluoride.[63][79][183]
6 transformers at a Russian railway
At least 17,000 metric tons of fluorine are produced each year. It costs only $5–8 per kilogram as uranium or sulfur hexafluoride, but many times more as an element because of handling challenges. Most processes using free fluorine in large amounts employ in situ generation under vertical integration.[184]
The largest application of fluorine gas, consuming up to 7,000 metric tons annually, is in the preparation of UF
6 for the nuclear fuel cycle. Fluorine is used to fluorinate uranium tetrafluoride, itself formed from uranium dioxide and hydrofluoric acid.[184] Fluorine is monoisotopic, so any mass differences between UF
6 molecules are due to the presence of 235
U or 238
U, enabling uranium enrichment via gaseous diffusion or gas centrifuge.[4][63] About 6,000 metric tons per year go into producing the inert dielectric SF
6 for high-voltage transformers and circuit breakers, eliminating the need for hazardous polychlorinated biphenyls associated with oil-filled devices.[185] Several fluorine compounds are used in electronics: rhenium and tungsten hexafluoride in chemical vapor deposition, tetrafluoromethane in plasma etching[186][187][188] and nitrogen trifluoride in cleaning equipment.[63] Fluorine is also used in the synthesis of organic fluorides, but its reactivity often necessitates conversion first to the gentler ClF
3, BrF
3, or IF
5, which together allow calibrated fluorination. Fluorinated pharmaceuticals use sulfur tetrafluoride instead.[63]
Inorganic fluorides
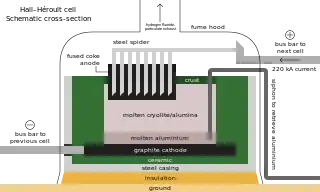
As with other iron alloys, around 3 kg (6.5 lb) metspar is added to each metric ton of steel; the fluoride ions lower its melting point and viscosity.[63][189] Alongside its role as an additive in materials like enamels and welding rod coats, most acidspar is reacted with sulfuric acid to form hydrofluoric acid, which is used in steel pickling, glass etching and alkane cracking.[63] One-third of HF goes into synthesizing cryolite and aluminium trifluoride, both fluxes in the Hall–Héroult process for aluminium extraction; replenishment is necessitated by their occasional reactions with the smelting apparatus. Each metric ton of aluminium requires about 23 kg (51 lb) of flux.[63][190] Fluorosilicates consume the second largest portion, with sodium fluorosilicate used in water fluoridation and laundry effluent treatment, and as an intermediate en route to cryolite and silicon tetrafluoride.[191] Other important inorganic fluorides include those of cobalt, nickel, and ammonium.[63][102][192]
Organic fluorides
Organofluorides consume over 20% of mined fluorite and over 40% of hydrofluoric acid, with refrigerant gases dominating and fluoropolymers increasing their market share.[63][193] Surfactants are a minor application but generate over $1 billion in annual revenue.[194] Due to the danger from direct hydrocarbon–fluorine reactions above −150 °C (−240 °F), industrial fluorocarbon production is indirect, mostly through halogen exchange reactions such as Swarts fluorination, in which chlorocarbon chlorines are substituted for fluorines by hydrogen fluoride under catalysts. Electrochemical fluorination subjects hydrocarbons to electrolysis in hydrogen fluoride, and the Fowler process treats them with solid fluorine carriers like cobalt trifluoride.[91][195]
Refrigerant gases
Halogenated refrigerants, termed Freons in informal contexts,[note 18] are identified by R-numbers that denote the amount of fluorine, chlorine, carbon, and hydrogen present.[63][196] Chlorofluorocarbons (CFCs) like R-11, R-12, and R-114 once dominated organofluorines, peaking in production in the 1980s. Used for air conditioning systems, propellants and solvents, their production was below one-tenth of this peak by the early 2000s, after widespread international prohibition.[63] Hydrochlorofluorocarbons (HCFCs) and hydrofluorocarbons (HFCs) were designed as replacements; their synthesis consumes more than 90% of the fluorine in the organic industry. Important HCFCs include R-22, chlorodifluoromethane, and R-141b. The main HFC is R-134a[63] with a new type of molecule HFO-1234yf, a Hydrofluoroolefin (HFO) coming to prominence owing to its global warming potential of less than 1% that of HFC-134a.[197]
Polymers

About 180,000 metric tons of fluoropolymers were produced in 2006 and 2007, generating over $3.5 billion revenue per year.[198] The global market was estimated at just under $6 billion in 2011 and was predicted to grow by 6.5% per year up to 2016.[199] Fluoropolymers can only be formed by polymerizing free radicals.[163]
Polytetrafluoroethylene (PTFE), sometimes called by its DuPont name Teflon,[200] represents 60–80% by mass of the world's fluoropolymer production.[198] The largest application is in electrical insulation since PTFE is an excellent dielectric. It is also used in the chemical industry where corrosion resistance is needed, in coating pipes, tubing, and gaskets. Another major use is in PFTE-coated fiberglass cloth for stadium roofs. The major consumer application is for non-stick cookware.[200] Jerked PTFE film becomes expanded PTFE (ePTFE), a fine-pored membrane sometimes referred to by the brand name Gore-Tex and used for rainwear, protective apparel, and filters; ePTFE fibers may be made into seals and dust filters.[200] Other fluoropolymers, including fluorinated ethylene propylene, mimic PTFE's properties and can substitute for it; they are more moldable, but also more costly and have lower thermal stability. Films from two different fluoropolymers replace glass in solar cells.[200][201]
The chemically resistant (but expensive) fluorinated ionomers are used as electrochemical cell membranes, of which the first and most prominent example is Nafion. Developed in the 1960s, it was initially deployed as fuel cell material in spacecraft and then replaced mercury-based chloralkali process cells. Recently, the fuel cell application has reemerged with efforts to install proton exchange membrane fuel cells into automobiles.[202][203][204] Fluoroelastomers such as Viton are crosslinked fluoropolymer mixtures mainly used in O-rings;[200] perfluorobutane (C4F10) is used as a fire-extinguishing agent.[205]
Surfactants
Fluorosurfactants are small organofluorine molecules used for repelling water and stains. Although expensive (comparable to pharmaceuticals at $200–2000 per kilogram), they yielded over $1 billion in annual revenues by 2006; Scotchgard alone generated over $300 million in 2000.[194][206][207] Fluorosurfactants are a minority in the overall surfactant market, most of which is taken up by much cheaper hydrocarbon-based products. Applications in paints are burdened by compounding costs; this use was valued at only $100 million in 2006.[194]
Agrichemicals
About 30% of agrichemicals contain fluorine,[208] most of them herbicides and fungicides with a few crop regulators. Fluorine substitution, usually of a single atom or at most a trifluoromethyl group, is a robust modification with effects analogous to fluorinated pharmaceuticals: increased biological stay time, membrane crossing, and altering of molecular recognition.[209] Trifluralin is a prominent example, with large-scale use in the U.S. as a weedkiller,[209][210] but it is a suspected carcinogen and has been banned in many European countries.[211] Sodium monofluoroacetate (1080) is a mammalian poison in which two acetic acid hydrogens are replaced with fluorine and sodium; it disrupts cell metabolism by replacing acetate in the citric acid cycle. First synthesized in the late 19th century, it was recognized as an insecticide in the early 20th, and was later deployed in its current use. New Zealand, the largest consumer of 1080, uses it to protect kiwis from the invasive Australian common brushtail possum.[212] Europe and the U.S. have banned 1080.[213][214][note 19]
Medicinal applications
Dental care
_gives_a_Fluoride_treatment_to_a_patient_during_a_Continuing_Promise_2009_medical_civil_service_projec.jpg.webp)
Population studies from the mid-20th century onwards show topical fluoride reduces dental caries. This was first attributed to the conversion of tooth enamel hydroxyapatite into the more durable fluorapatite, but studies on pre-fluoridated teeth refuted this hypothesis, and current theories involve fluoride aiding enamel growth in small caries.[215] After studies of children in areas where fluoride was naturally present in drinking water, controlled public water supply fluoridation to fight tooth decay[216] began in the 1940s and is now applied to water supplying 6 percent of the global population, including two-thirds of Americans.[217][218] Reviews of the scholarly literature in 2000 and 2007 associated water fluoridation with a significant reduction of tooth decay in children.[219] Despite such endorsements and evidence of no adverse effects other than mostly benign dental fluorosis,[220] opposition still exists on ethical and safety grounds.[218][221] The benefits of fluoridation have lessened, possibly due to other fluoride sources, but are still measurable in low-income groups.[222] Sodium monofluorophosphate and sometimes sodium or tin(II) fluoride are often found in fluoride toothpastes, first introduced in the U.S. in 1955 and now ubiquitous in developed countries, alongside fluoridated mouthwashes, gels, foams, and varnishes.[222][223]
Pharmaceuticals

Twenty percent of modern pharmaceuticals contain fluorine.[224] One of these, the cholesterol-reducer atorvastatin (Lipitor), made more revenue than any other drug until it became generic in 2011.[225] The combination asthma prescription Seretide, a top-ten revenue drug in the mid-2000s, contains two active ingredients, one of which – fluticasone – is fluorinated.[226] Many drugs are fluorinated to delay inactivation and lengthen dosage periods because the carbon–fluorine bond is very stable.[227] Fluorination also increases lipophilicity because the bond is more hydrophobic than the carbon–hydrogen bond, and this often helps in cell membrane penetration and hence bioavailability.[226]
Tricyclics and other pre-1980s antidepressants had several side effects due to their non-selective interference with neurotransmitters other than the serotonin target; the fluorinated fluoxetine was selective and one of the first to avoid this problem. Many current antidepressants receive this same treatment, including the selective serotonin reuptake inhibitors: citalopram, its isomer escitalopram, and fluvoxamine and paroxetine.[228][229] Quinolones are artificial broad-spectrum antibiotics that are often fluorinated to enhance their effects. These include ciprofloxacin and levofloxacin.[230][231][232][233] Fluorine also finds use in steroids:[234] fludrocortisone is a blood pressure-raising mineralocorticoid, and triamcinolone and dexamethasone are strong glucocorticoids.[235] The majority of inhaled anesthetics are heavily fluorinated; the prototype halothane is much more inert and potent than its contemporaries. Later compounds such as the fluorinated ethers sevoflurane and desflurane are better than halothane and are almost insoluble in blood, allowing faster waking times.[236][237]
PET scanning
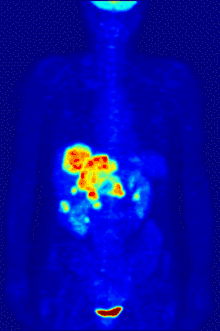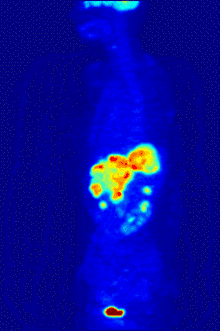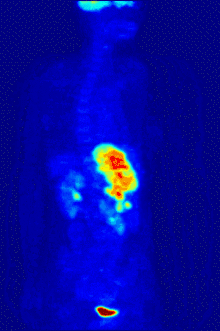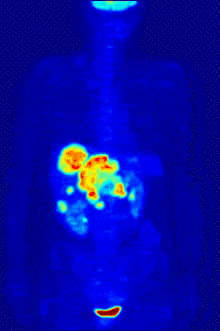
F PET scan with glucose tagged with radioactive fluorine-18. The normal brain and kidneys take up enough glucose to be imaged. A malignant tumor is seen in the upper abdomen. Radioactive fluorine is seen in urine in the bladder.
Fluorine-18 is often found in radioactive tracers for positron emission tomography, as its half-life of almost two hours is long enough to allow for its transport from production facilities to imaging centers.[238] The most common tracer is fluorodeoxyglucose[238] which, after intravenous injection, is taken up by glucose-requiring tissues such as the brain and most malignant tumors;[239] computer-assisted tomography can then be used for detailed imaging.[240]
Oxygen carriers
Liquid fluorocarbons can hold large volumes of oxygen or carbon dioxide, more so than blood, and have attracted attention for their possible uses in artificial blood and in liquid breathing.[241] Because fluorocarbons do not normally mix with water, they must be mixed into emulsions (small droplets of perfluorocarbon suspended in water) to be used as blood.[242][243] One such product, Oxycyte, has been through initial clinical trials.[244] These substances can aid endurance athletes and are banned from sports; one cyclist's near death in 1998 prompted an investigation into their abuse.[245][246] Applications of pure perfluorocarbon liquid breathing (which uses pure perfluorocarbon liquid, not a water emulsion) include assisting burn victims and premature babies with deficient lungs. Partial and complete lung filling have been considered, though only the former has had any significant tests in humans.[247] An Alliance Pharmaceuticals effort reached clinical trials but was abandoned because the results were not better than normal therapies.[248]
Biological role

Fluorine is not essential for humans and mammals, but small amounts are known to be beneficial for the strengthening of dental enamel (where the formation of fluorapatite makes the enamel more resistant to attack, from acids produced by bacterial fermentation of sugars). Small amounts of fluorine may be beneficial for bone strength, but the latter has not been definitively established.[249] Both the WHO[250] and the Institute of Medicine of the US National Academies[251] publish recommended daily allowance (RDA) and upper tolerated intake of fluorine, which varies with age and gender.
Natural organofluorines have been found in microorganisms and plants[66] but not animals.[252] The most common is fluoroacetate, which is used as a defense against herbivores by at least 40 plants in Africa, Australia and Brazil.[213] Other examples include terminally fluorinated fatty acids, fluoroacetone, and 2-fluorocitrate.[252] An enzyme that binds fluorine to carbon – adenosyl-fluoride synthase – was discovered in bacteria in 2002.[253]
Toxicity



.svg.png.webp)
Elemental fluorine is highly toxic to living organisms. Its effects in humans start at concentrations lower than hydrogen cyanide's 50 ppm[255] and are similar to those of chlorine:[256] significant irritation of the eyes and respiratory system as well as liver and kidney damage occur above 25 ppm, which is the immediately dangerous to life and health value for fluorine.[257] Eyes and noses are seriously damaged at 100 ppm,[257] and inhalation of 1,000 ppm fluorine will cause death in minutes,[258] compared to 270 ppm for hydrogen cyanide.[259]
| Hazards | |
|---|---|
| GHS pictograms |     |
| GHS Signal word | Danger |
| H270, H330, H314, H318[260] | |
| NFPA 704 (fire diamond) | |

Hydrofluoric acid
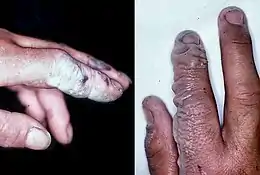
Hydrofluoric acid is the weakest of the hydrohalic acids, having a pKa of 3.2 at 25 °C.[262] It is a volatile liquid due to the presence of hydrogen bonding (while the other hydrohalic acids are gases). It is able to attack glass, concrete, metals, organic matter.[263]
Hydrofluoric acid is a contact poison with greater hazards than many strong acids like sulfuric acid even though it is weak: it remains neutral in aqueous solution and thus penetrates tissue faster, whether through inhalation, ingestion or the skin, and at least nine U.S. workers died in such accidents from 1984 to 1994. It reacts with calcium and magnesium in the blood leading to hypocalcemia and possible death through cardiac arrhythmia.[264] Insoluble calcium fluoride formation triggers strong pain[265] and burns larger than 160 cm2 (25 in2) can cause serious systemic toxicity.[266]
Exposure may not be evident for eight hours for 50% HF, rising to 24 hours for lower concentrations, and a burn may initially be painless as hydrogen fluoride affects nerve function. If skin has been exposed to HF, damage can be reduced by rinsing it under a jet of water for 10–15 minutes and removing contaminated clothing.[267] Calcium gluconate is often applied next, providing calcium ions to bind with fluoride; skin burns can be treated with 2.5% calcium gluconate gel or special rinsing solutions.[268][269][270] Hydrofluoric acid absorption requires further medical treatment; calcium gluconate may be injected or administered intravenously. Using calcium chloride – a common laboratory reagent – in lieu of calcium gluconate is contraindicated, and may lead to severe complications. Excision or amputation of affected parts may be required.[266][271]
Fluoride ion
Soluble fluorides are moderately toxic: 5–10 g sodium fluoride, or 32–64 mg fluoride ions per kilogram of body mass, represents a lethal dose for adults.[272] One-fifth of the lethal dose can cause adverse health effects,[273] and chronic excess consumption may lead to skeletal fluorosis, which affects millions in Asia and Africa.[273][274] Ingested fluoride forms hydrofluoric acid in the stomach which is easily absorbed by the intestines, where it crosses cell membranes, binds with calcium and interferes with various enzymes, before urinary excretion. Exposure limits are determined by urine testing of the body's ability to clear fluoride ions.[273][275]
Historically, most cases of fluoride poisoning have been caused by accidental ingestion of insecticides containing inorganic fluorides.[276] Most current calls to poison control centers for possible fluoride poisoning come from the ingestion of fluoride-containing toothpaste.[273] Malfunctioning water fluoridation equipment is another cause: one incident in Alaska affected almost 300 people and killed one person.[277] Dangers from toothpaste are aggravated for small children, and the Centers for Disease Control and Prevention recommends supervising children below six brushing their teeth so that they do not swallow toothpaste.[278] One regional study examined a year of pre-teen fluoride poisoning reports totaling 87 cases, including one death from ingesting insecticide. Most had no symptoms, but about 30% had stomach pains.[276] A larger study across the U.S. had similar findings: 80% of cases involved children under six, and there were few serious cases.[279]
Environmental concerns
Atmosphere
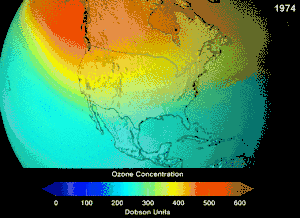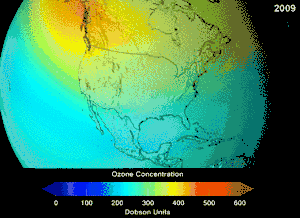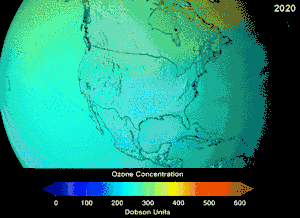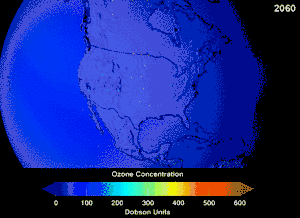
The Montreal Protocol, signed in 1987, set strict regulations on chlorofluorocarbons (CFCs) and bromofluorocarbons due to their ozone damaging potential (ODP). The high stability which suited them to their original applications also meant that they were not decomposing until they reached higher altitudes, where liberated chlorine and bromine atoms attacked ozone molecules.[281] Even with the ban, and early indications of its efficacy, predictions warned that several generations would pass before full recovery.[282][283] With one-tenth the ODP of CFCs, hydrochlorofluorocarbons (HCFCs) are the current replacements,[284] and are themselves scheduled for substitution by 2030–2040 by hydrofluorocarbons (HFCs) with no chlorine and zero ODP.[285] In 2007 this date was brought forward to 2020 for developed countries;[286] the Environmental Protection Agency had already prohibited one HCFC's production and capped those of two others in 2003.[285] Fluorocarbon gases are generally greenhouse gases with global-warming potentials (GWPs) of about 100 to 10,000; sulfur hexafluoride has a value of around 20,000.[287] An outlier is HFO-1234yf which is a new type of refrigerant called a Hydrofluoroolefin (HFO) and has attracted global demand due to its GWP of less than 1 compared to 1,430 for the current refrigerant standard HFC-134a.[197]
Biopersistence
Organofluorines exhibit biopersistence due to the strength of the carbon–fluorine bond. Perfluoroalkyl acids (PFAAs), which are sparingly water-soluble owing to their acidic functional groups, are noted persistent organic pollutants;[289] perfluorooctanesulfonic acid (PFOS) and perfluorooctanoic acid (PFOA) are most often researched.[290][291][292] PFAAs have been found in trace quantities worldwide from polar bears to humans, with PFOS and PFOA known to reside in breast milk and the blood of newborn babies. A 2013 review showed a slight correlation between groundwater and soil PFAA levels and human activity; there was no clear pattern of one chemical dominating, and higher amounts of PFOS were correlated to higher amounts of PFOA.[290][291][293] In the body, PFAAs bind to proteins such as serum albumin; they tend to concentrate within humans in the liver and blood before excretion through the kidneys. Dwell time in the body varies greatly by species, with half-lives of days in rodents, and years in humans.[290][291][294] High doses of PFOS and PFOA cause cancer and death in newborn rodents but human studies have not established an effect at current exposure levels.[290][291][294]
See also
- Argon fluoride laser
- Electrophilic fluorination
- Fluoride selective electrode, which measures fluoride concentration
- Fluorine absorption dating
- Fluorous chemistry, a process used to separate reagents from organic solvents
- Krypton fluoride laser
- Radical fluorination
Notes
- Sources disagree on the radii of oxygen, fluorine, and neon atoms Precise comparison is thus impossible.
- α-Fluorine has a regular pattern of molecules and is a crystalline solid, but its molecules do not have a specific orientation. β-Fluorine's molecules have fixed locations and minimal rotational uncertainty. For further detail on α-fluorine, see the 1970 structure by Pauling.[43] For further detail on the concept of disorder in crystals, see the referenced general reviews.[44][45]
- A loud click is heard. Samples may shatter and sample windows blow out.
- The ratio of the angular momentum to magnetic moment is called the gyromagnetic ratio. "Certain nuclei can for many purposes be thought of as spinning round an axis like the Earth or like a top. In general the spin endows them with angular momentum and with a magnetic moment; the first because of their mass, the second because all or part of their electric charge may be rotating with the mass."[49]
- Basilius Valentinus supposedly described fluorite in the late 15th century, but because his writings were uncovered 200 years later, this work's veracity is doubtful.[71][72][73]
- Or perhaps from as early as 1670 onwards; Partington[77] and Weeks[76] give differing accounts.
- Fl, since 2012, is used for flerovium.
- Davy, Gay-Lussac, Thénard, and the Irish chemists Thomas and George Knox were injured. Belgian chemist Paulin Louyet and French chemist Jérôme Nicklès died. Moissan also experienced serious hydrogen fluoride poisoning.[76][86]
- Also honored was his invention of the electric arc furnace.
- Fluorine in F
2 is defined to have oxidation state 0. The unstable species F−
2 and F−
3, which decompose at around 40 K, have intermediate oxidation states;[97] F+
4 and a few related species are predicted to be stable.[98] - The metastable boron and nitrogen monofluoride have higher-order fluorine bonds, and some metal complexes use it as a bridging ligand. Hydrogen bonding is another possibility.
- ZrF
4 melts at 932 °C (1710 °F),[111] HfF
4 sublimes at 968 °C (1774 °F),[108] and UF
4 melts at 1036 °C (1897 °F).[112] - These thirteen are those of molybdenum, technetium, ruthenium, rhodium, tungsten, rhenium, osmium, iridium, platinum, polonium, uranium, neptunium, and plutonium.
- See also the explanation by Clark.[130]
- Carbon tetrafluoride is formally organic, but is included here rather than in the organofluorine chemistry section – where more complex carbon-fluorine compounds are discussed – for comparison with SiF
4 and GeF
4. - Perfluorocarbon and fluorocarbon are IUPAC synonyms for molecules containing carbon and fluorine only, but in colloquial and commercial contexts the latter term may refer to any carbon- and fluorine-containing molecule, possibly with other elements.
- This terminology is imprecise, and perfluorinated substance is also used.[160]
- This DuPont trademark is sometimes further misused for CFCs, HFCs, or HCFCs.
- American sheep and cattle collars may use 1080 against predators like coyotes.
Sources
Citations
- Meija, Juris; et al. (2016). "Atomic weights of the elements 2013 (IUPAC Technical Report)". Pure and Applied Chemistry. 88 (3): 265–91. doi:10.1515/pac-2015-0305.
- Jaccaud et al. 2000, p. 381.
- Haynes 2011, p. 4.121.
- Jaccaud et al. 2000, p. 382.
- Compressed Gas Association 1999, p. 365.
- Dean 1999, p. 4.6.
- Dean 1999, p. 4.35.
- Matsui 2006, p. 257.
- Yaws & Braker 2001, p. 385.
- Mackay, Mackay & Henderson 2002, p. 72.
- Cheng et al. 1999.
- Chisté & Bé 2011.
- Lee, Stephen; et al. (2014). "Monofluoroacetate-Containing Plants That Are Potentially Toxic to Livestock". Journal of Agricultural and Food Chemistry. ACS Publications. 62 (30): 7345–7354. doi:10.1021/jf500563h. PMID 24724702.
- Dean 1999, p. 564.
- Lide 2004, pp. 10.137–10.138.
- Moore, Stanitski & Jurs 2010, p. 156.
- Cordero et al. 2008.
- Pyykkö & Atsumi 2009.
- Greenwood & Earnshaw 1998, p. 804.
- Macomber 1996, p. 230
- Nelson 1947.
- Lidin, Molochko & Andreeva 2000, pp. 442–455.
- Wiberg, Wiberg & Holleman 2001, p. 404.
- Patnaik 2007, p. 472.
- Aigueperse et al. 2000, p. 400.
- Greenwood & Earnshaw 1998, pp. 76, 804.
- Kuriakose & Margrave 1965.
- Hasegawa et al. 2007.
- Lagow 1970, pp. 64–78.
- Lidin, Molochko & Andreeva 2000, p. 252.
- Tanner Industries 2011.
- Morrow, Perry & Cohen 1959.
- Emeléus & Sharpe 1974, p. 111.
- Wiberg, Wiberg & Holleman 2001, p. 457.
- Brantley 1949, p. 26.
- Jaccaud et al. 2000, p. 383.
- Pitzer 1975.
- Khriachtchev et al. 2000.
- Burdon, Emson & Edwards 1987.
- Lide 2004, p. 4.12.
- Dean 1999, p. 523.
- Pauling, Keaveny & Robinson 1970.
- Bürgi 2000.
- Müller 2009.
- Young 1975, p. 10.
- Barrett, Meyer & Wasserman 1967.
- National Nuclear Data Center & NuDat 2.1, Fluorine-19.
- Vigoureux 1961.
- Meusinger, Chippendale & Fairhurst 2012, pp. 752, 754.
- National Nuclear Data Center & NuDat 2.1.
- NUBASE 2016, pp. 030001-23–030001-27.
- NUBASE 2016, pp. 030001–24.
- Cameron 1973.
- Croswell 2003.
- Clayton 2003, pp. 101–104.
- Renda et al. 2004.
- Jaccaud et al. 2000, p. 384.
- Schulze-Makuch & Irwin 2008, p. 121.
- Haxel, Hedrick & Orris 2005.
- Greenwood & Earnshaw 1998, p. 795.
- Norwood & Fohs 1907, p. 52.
- Villalba, Ayres & Schroder 2008.
- Kelly & Miller 2005.
- Lusty et al. 2008.
- Gribble 2002.
- Richter, Hahn & Fuchs 2001, p. 3.
- Schmedt, Mangstl & Kraus 2012.
- Greenwood & Earnshaw 1998, p. 790.
- Senning 2007, p. 149.
- Stillman 1912.
- Principe 2012, pp. 140, 145.
- Agricola, Hoover & Hoover 1912, footnotes and commentary, pp. xxx, 38, 409, 430, 461, 608.
- Greenwood & Earnshaw 1998, p. 109.
- Agricola, Hoover & Hoover 1912, preface, pp. 380–381.
- Weeks 1932.
- Partington 1923.
- Marggraf 1770.
- Kirsch 2004, pp. 3–10.
- Scheele 1771.
- Ampère 1816.
- https://books.google.com/books?id=kslaDwAAQBAJ&pg=PA3
- Davy 1813, p. 278.
- Banks 1986, p. 11.
- Storer 1864, pp. 278–280.
- Toon 2011.
- Asimov 1966, p. 162.
- Greenwood & Earnshaw 1998, pp. 789–791.
- Moissan 1886.
- Viel & Goldwhite 1993, p. 35.
- Okazoe 2009.
- Hounshell & Smith 1988, pp. 156–157.
- DuPont 2013a.
- Meyer 1977, p. 111.
- Kirsch 2004, pp. 60–66.
- Riedel & Kaupp 2009.
- Wiberg, Wiberg & Holleman 2001, p. 422.
- Schlöder & Riedel 2012.
- Harbison 2002.
- Edwards 1994, p. 515.
- Katakuse et al. 1999, p. 267.
- Aigueperse et al. 2000, pp. 420–422.
- Walsh 2009, pp. 99–102, 118–119.
- Emeléus & Sharpe 1983, pp. 89–97.
- Babel & Tressaud 1985, pp. 91–96.
- Einstein et al. 1967.
- Brown et al. 2005, p. 144.
- Perry 2011, p. 193.
- Kern et al. 1994.
- Lide 2004, pp. 4.60, 4.76, 4.92, 4.96.
- Lide 2004, p. 4.96.
- Lide 2004, p. 4.92.
- Greenwood & Earnshaw 1998, p. 964.
- Becker & Müller 1990.
- Greenwood & Earnshaw 1998, p. 990.
- Lide 2004, pp. 4.72, 4.91, 4.93.
- Greenwood & Earnshaw 1998, pp. 561–563.
- Emeléus & Sharpe 1983, pp. 256–277.
- Mackay, Mackay & Henderson 2002, pp. 355–356.
- Greenwood & Earnshaw 1998, (various pages, by metal in respective chapters).
- Lide 2004, pp. 4.71, 4.78, 4.92.
- Drews et al. 2006.
- Greenwood & Earnshaw 1998, p. 819.
- Bartlett 1962.
- Pauling 1960, pp. 454–464.
- Atkins & Jones 2007, pp. 184–185.
- Emsley 1981.
- Greenwood & Earnshaw 1998, pp. 812–816.
- Wiberg, Wiberg & Holleman 2001, p. 425.
- Clark 2002.
- Chambers & Holliday 1975, pp. 328–329.
- Air Products and Chemicals 2004, p. 1.
- Noury, Silvi & Gillespie 2002.
- Chang & Goldsby 2013, p. 706.
- Ellis 2001, p. 69.
- Aigueperse et al. 2000, p. 423.
- Wiberg, Wiberg & Holleman 2001, p. 897.
- Raghavan 1998, pp. 164–165.
- Godfrey et al. 1998, p. 98.
- Aigueperse et al. 2000, p. 432.
- Murthy, Mehdi Ali & Ashok 1995, pp. 180–182, 206–208.
- Greenwood & Earnshaw 1998, pp. 638–640, 683–689, 767–778.
- Wiberg, Wiberg & Holleman 2001, pp. 435–436.
- Greenwood & Earnshaw 1998, pp. 828–830.
- Patnaik 2007, pp. 478–479.
- Moeller, Bailar & Kleinberg 1980, p. 236.
- Wiberg, Wiberg & Holleman 2001, pp. 392–393.
- Wiberg, Wiberg & Holleman 2001, p. 395–397, 400.
- Lewars 2008, p. 68.
- Pitzer 1993, p. 111.
- Lewars 2008, p. 67.
- Bihary, Chaban & Gerber 2002.
- Lewars 2008, p. 71.
- Hoogers 2002, pp. 4–12.
- O'Hagan 2008.
- Siegemund et al. 2005, p. 444.
- Sandford 2000, p. 455.
- Siegemund et al. 2005, pp. 451–452.
- Barbee, McCormack & Vartanian 2000, p. 116.
- Posner et al. 2013, pp. 187–190.
- Posner 2011, p. 27.
- Salager 2002, p. 45.
- Carlson & Schmiegel 2000, p. 3.
- Carlson & Schmiegel 2000, pp. 3–4.
- Rhoades 2008, p. 2.
- Okada et al. 1998.
- Carlson & Schmiegel 2000, p. 4.
- Aigueperse et al. 2000.
- Norris Shreve; Joseph Brink, Jr. (1977). Chemical Process Industries (4 ed.). p. 321. ISBN 0070571457.
- Jaccaud et al. 2000, p. 386.
- Jaccaud et al. 2000, pp. 384–285.
- Greenwood & Earnshaw 1998, pp. 796–797.
- Jaccaud et al. 2000, pp. 384–385.
- Jaccaud et al. 2000, pp. 390–391.
- Shriver & Atkins 2010, p. 427.
- Christe 1986.
- Christe Research Group n.d.
- Carey 2008, p. 173.
- Miller 2003b.
- PRWeb 2012.
- Bombourg 2012.
- TMR 2013.
- Fulton & Miller 2006, p. 471.
- Jaccaud et al. 2000, p. 392.
- Aigueperse et al. 2000, p. 430.
- Jaccaud et al. 2000, pp. 391–392.
- El-Kareh 1994, p. 317.
- Arana et al. 2007.
- Miller 2003a.
- Energetics, Inc. 1997, pp. 41, 50.
- Aigueperse et al. 2000, p. 428.
- Willey 2007, p. 113.
- PRWeb 2010.
- Renner 2006.
- Green et al. 1994, pp. 91–93.
- DuPont 2013b.
- Walter 2013.
- Buznik 2009.
- PRWeb 2013.
- Martin 2007, pp. 187–194.
- DeBergalis 2004.
- Grot 2011, pp. 1–10.
- Ramkumar 2012, p. 567.
- Burney 1999, p. 111.
- Slye 2012, p. 10.
- Kissa 2001, pp. 516–551.
- Ullmann 2008, pp. 538, 543–547.
- ICIS 2006.
- Theodoridis 2006.
- EPA 1996.
- DG Environment 2007.
- Beasley 2002.
- Proudfoot, Bradberry & Vale 2006.
- Eisler 1995.
- Pizzo 2007.
- CDC 2001.
- Ripa 1993.
- Cheng, Chalmers & Sheldon 2007.
- NHMRC 2007; see Yeung 2008 for a summary.
- Marya 2011, p. 343.
- Armfield 2007.
- Baelum, Sheiham & Burt 2008, p. 518.
- Cracher 2012, p. 12.
- Emsley 2011, p. 178.
- Johnson 2011.
- Swinson 2005.
- Hagmann 2008.
- Mitchell 2004, pp. 37–39.
- Preskorn 1996, chap. 2.
- Werner et al. 2011.
- Brody 2012.
- Nelson et al. 2007.
- King, Malone & Lilley 2000.
- Parente 2001, p. 40.
- Raj & Erdine 2012, p. 58.
- Filler & Saha 2009.
- Bégué & Bonnet-Delpon 2008, pp. 335–336.
- Schmitz et al. 2000.
- Bustamante & Pedersen 1977.
- Alavi & Huang 2007, p. 41.
- Gabriel et al. 1996.
- Sarkar 2008.
- Schimmeyer 2002.
- Davis 2006.
- Gains 1998.
- Taber 1999.
- Shaffer, Wolfson & Clark Jr 1992, p. 102.
- Kacmarek et al. 2006.
- Nielsen 2009.
- Olivares & Uauy 2004
- "Dietary Reference Intakes (DRIs): Recommended Dietary Allowances and Adequate Intakes, Elements" (PDF). Food and Nutrition Board, Institute of Medicine, National Academies. Archived from the original (PDF) on 13 November 2018. Retrieved 2 January 2019.
- Murphy, Schaffrath & O'Hagan 2003
- O'Hagan et al. 2002.
- National Oceanic and Atmospheric Administration.
- The National Institute for Occupational Safety and Health 1994a.
- The National Institute for Occupational Safety and Health 1994b.
- Keplinger & Suissa 1968.
- Emsley 2011, p. 179.
- Biller 2007, p. 939.
- "Fluorine. Safety data sheet" (PDF). Airgas. Archived from the original (PDF) on 19 April 2015.
- Eaton 1997.
- "Inorganic Chemistry" by Gary L. Miessler and Donald A. Tarr, 4th edition, Pearson
- "Inorganic Chemistry" by Shriver, Weller, Overton, Rourke and Armstrong, 6th edition, Freeman
- Blodgett, Suruda & Crouch 2001.
- Hoffman et al. 2007, p. 1333.
- HSM 2006.
- Fischman 2001, pp. 458–459.
- El Saadi et al. 1989.
- Roblin et al. 2006.
- Hultén et al. 2004.
- Zorich 1991, pp. 182–183.
- Liteplo et al. 2002, p. 100.
- Shin & Silverberg 2013.
- Reddy 2009.
- Baez, Baez & Marthaler 2000.
- Augenstein et al. 1991.
- Gessner et al. 1994.
- CDC 2013.
- Shulman & Wells 1997.
- Beck et al. 2011.
- Aucamp & Björn 2010, pp. 4–6, 41, 46–47.
- Mitchell Crow 2011.
- Barry & Phillips 2006.
- EPA 2013a.
- EPA 2013b.
- McCoy 2007.
- Forster et al. 2007, pp. 212–213.
- Schwarcz 2004, p. 37.
- Giesy & Kannan 2002.
- Steenland, Fletcher & Savitz 2010.
- Betts 2007.
- EPA 2012.
- Zareitalabad et al. 2013.
- Lau et al. 2007.
Indexed references
- Agricola, Georgius; Hoover, Herbert Clark; Hoover, Lou Henry (1912). De Re Metallica. London: The Mining Magazine.
- Aigueperse, J.; Mollard, P.; Devilliers, D.; Chemla, M.; Faron, R.; Romano, R. E.; Cue, J. P. (2000). "Fluorine Compounds, Inorganic". Ullmann's Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH. pp. 397–441. doi:10.1002/14356007.
- Air Products and Chemicals (2004). "Safetygram #39 Chlorine Trifluoride" (PDF). Air Products and Chemicals. Archived from the original (PDF) on 18 March 2006. Retrieved 16 February 2014.
- Alavi, Abbas; Huang, Steve S. (2007). "Positron Emission Tomography in Medicine: An Overview". In Hayat, M. A. (ed.). Cancer Imaging, Volume 1: Lung and Breast Carcinomas. Burlington: Academic Press. pp. 39–44. ISBN 978-0-12-370468-9.
- Ampère, André-Marie (1816). "Suite d'une classification naturelle pour les corps simples". Annales de chimie et de physique (in French). 2: 1–5.
- Arana, L. R.; Mas, N.; Schmidt, R.; Franz, A. J.; Schmidt, M. A.; Jensen, K. F. (2007). "Isotropic Etching of Silicon in Fluorine Gas for MEMS Micromachining". Journal of Micromechanics and Microengineering. 17 (2): 384–392. Bibcode:2007JMiMi..17..384A. doi:10.1088/0960-1317/17/2/026.
- Armfield, J. M. (2007). "When Public Action Undermines Public Health: A Critical Examination of Antifluoridationist Literature". Australia and New Zealand Health Policy. 4: 25. doi:10.1186/1743-8462-4-25. PMC 2222595. PMID 18067684.
- Asimov, Isaac (1966). The Noble Gases. New York: Basic Books. ISBN 978-0-465-05129-8.
- Atkins, Peter; Jones, Loretta (2007). Chemical Principles: The Quest for Insight (4th ed.). New York: W. H. Freeman. ISBN 978-1-4292-0965-6.
- Aucamp, Pieter J.; Björn, Lars Olof (2010). "Questions and Answers about the Environmental Effects of the Ozone Layer Depletion and Climate Change: 2010 Update" (PDF). United Nations Environmental Programme. Archived from the original (PDF) on 3 September 2013. Retrieved 14 October 2013.
- Audi, G.; Kondev, F. G.; Wang, M.; Huang, W. J.; Naimi, S. (2017). "The NUBASE2016 evaluation of nuclear properties" (PDF). Chinese Physics C. 41 (3): 030001. Bibcode:2017ChPhC..41c0001A. doi:10.1088/1674-1137/41/3/030001..
- Augenstein, W. L.; et al. (1991). "Fluoride Ingestion in Children: A Review of 87 cases". Pediatrics. 88 (5): 907–912. PMID 1945630.
- Babel, Dietrich; Tressaud, Alain (1985). "Crystal Chemistry of Fluorides". In Hagenmuller, Paul (ed.). Inorganic Solid Fluorides: Chemistry And Physics. Orlando: Academic Press. pp. 78–203. ISBN 978-0-12-412490-5.
- Baelum, Vibeke; Sheiham, Aubrey; Burt, Brian (2008). "Caries Control for Populations". In Fejerskov, Ole; Kidd, Edwina (eds.). Dental Caries: The Disease and Its Clinical Management (2nd ed.). Oxford: Blackwell Munksgaard. pp. 505–526. ISBN 978-1-4051-3889-5.
- Baez, Ramon J.; Baez, Martha X.; Marthaler, Thomas M. (2000). "Urinary Fluoride Excretion by Children 4–6 Years Old in a South Texas Community". Revista Panamericana de Salud Pública. 7 (4): 242–248. doi:10.1590/S1020-49892000000400005. PMID 10846927.
- Banks, R. E. (1986). "Isolation of fluorine by Moissan: Setting the scene". Journal of Fluorine Chemistry. 33 (1–4): 3–26. doi:10.1016/S0022-1139(00)85269-0.
- Barbee, K.; McCormack, K.; Vartanian, V. (2000). "EHS Concerns with Ozonated Water Spray Processing". In Mendicino, L. (ed.). Environmental Issues in the Electronics and Semiconductor Industries. Pennington, NJ: The Electrochemical Society. pp. 108–121. ISBN 978-1-56677-230-3.
- Barrett, C. S.; Meyer, L.; Wasserman, J. (1967). "Argon—Fluorine Phase Diagram". The Journal of Chemical Physics. 47 (2): 740–743. Bibcode:1967JChPh..47..740B. doi:10.1063/1.1711946.
- Barry, Patrick L.; Phillips, Tony (26 May 2006). "Good News and a Puzzle". National Aeronautics and Space Administration. Retrieved 6 January 2012.
- Bartlett, N. (1962). "Xenon Hexafluoroplatinate (V) Xe+[PtF6]−". Proceedings of the Chemical Society (6): 218. doi:10.1039/PS9620000197.
- Beasley, Michael (August 2002). Guidelines for the safe use of sodium fluoroacetate (1080) (PDF). Wellington: Occupational Safety & Health Service, Department of Labour (New Zealand). ISBN 0-477-03664-3. Archived from the original (PDF) on 11 November 2013. Retrieved 11 November 2013.
- Beck, Jefferson; Newman, Paul; Schindler, Trent L.; Perkins, Lori (2011). "What Would have Happened to the Ozone Layer if Chlorofluorocarbons (CFCs) had not been Regulated?". National Aeronautics and Space Administration. Retrieved 15 October 2013.
- Becker, S.; Müller, B. G. (1990). "Vanadium Tetrafluoride". Angewandte Chemie International Edition in English. 29 (4): 406–407. doi:10.1002/anie.199004061.
- Bégué, Jean-Pierre; Bonnet-Delpon, Danièle (2008). Bioorganic and Medicinal Chemistry of Fluorine. Hoboken: John Wiley & Sons. ISBN 978-0-470-27830-7.
- Betts, K. S. (2007). "Perfluoroalkyl Acids: What is the Evidence Telling Us?". Environmental Health Perspectives. 115 (5): A250–A256. doi:10.1289/ehp.115-a250. PMC 1867999. PMID 17520044.
- Bihary, Z.; Chaban, G. M.; Gerber, R. B. (2002). "Stability of a Chemically Bound Helium Compound in High-pressure Solid Helium". The Journal of Chemical Physics. 117 (11): 5105–5108. Bibcode:2002JChPh.117.5105B. doi:10.1063/1.1506150.
- Biller, José (2007). Interface of Neurology and Internal Medicine (illustrated ed.). Philadelphia: Lippincott Williams & Wilkins. ISBN 978-0-7817-7906-7.
- Blodgett, D. W.; Suruda, A. J.; Crouch, B. I. (2001). "Fatal Unintentional Occupational Poisonings by Hydrofluoric Acid in the U.S" (PDF). American Journal of Industrial Medicine. 40 (2): 215–220. doi:10.1002/ajim.1090. PMID 11494350. Archived from the original (PDF) on 17 July 2012.
- Bombourg, Nicolas (4 July 2012). "World Fluorochemicals Market, Freedonia". Reporterlinker. Retrieved 20 October 2013.
- Brantley, L. R. (1949). Squires, Roy; Clarke, Arthur C. (eds.). "Fluorine". Pacific Rockets: Journal of the Pacific Rocket Society. South Pasadena: Sawyer Publishing/Pacific Rocket Society Historical Library. 3 (1): 11–18. ISBN 978-0-9794418-5-1.
- Brody, Jane E. (10 September 2012). "Popular Antibiotics May Carry Serious Side Effects". The New York Times Well Blog. Retrieved 18 October 2013.
- Brown, Paul L.; Mompean, Federico J.; Perrone, Jane; Illemassène, Myriam (2005). Chemical Thermodynamics of Zirconium. Amsterdam: Elsevier B. V. ISBN 978-0-444-51803-3.
- Burdon, J.; Emson, B.; Edwards, A. J. (1987). "Is Fluorine Gas Really Yellow?". Journal of Fluorine Chemistry. 34 (3–4): 471–474. doi:10.1016/S0022-1139(00)85188-X.
- Bürgi, H. B. (2000). "Motion and Disorder in Crystal Structure Analysis: Measuring and Distinguishing them". Annual Review of Physical Chemistry. 51: 275–296. Bibcode:2000ARPC...51..275B. doi:10.1146/annurev.physchem.51.1.275. PMID 11031283.
- Burney, H. (1999). "Past, Present and Future of the Chlor-Alkali Industry". In Burney, H. S.; Furuya, N.; Hine, F.; Ota, K.-I. (eds.). Chlor-Alkali and Chlorate Technology: R. B. MacMullin Memorial Symposium. Pennington: The Electrochemical Society. pp. 105–126. ISBN 1-56677-244-3.
- Bustamante, E.; Pedersen, P. L. (1977). "High Aerobic Glycolysis of Rat Hepatoma Cells in Culture: Role of Mitochondrial Hexokinase". Proceedings of the National Academy of Sciences. 74 (9): 3735–3739. Bibcode:1977PNAS...74.3735B. doi:10.1073/pnas.74.9.3735. PMC 431708. PMID 198801.
- Buznik, V. M. (2009). "Fluoropolymer Chemistry in Russia: Current Situation and Prospects". Russian Journal of General Chemistry. 79 (3): 520–526. doi:10.1134/S1070363209030335. S2CID 97518401.
- Cameron, A. G. W. (1973). "Abundance of the Elements in the Solar System" (PDF). Space Science Reviews. 15 (1): 121–146. Bibcode:1973SSRv...15..121C. doi:10.1007/BF00172440. S2CID 120201972. Archived from the original (PDF) on 21 October 2011.
- Carey, Charles W. (2008). African Americans in Science. Santa Barbara: ABC-CLIO. ISBN 978-1-85109-998-6.
- Carlson, D. P.; Schmiegel, W. (2000). "Fluoropolymers, Organic". Ullmann's Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH. pp. 495–533. doi:10.1002/14356007.a11_393.
- Centers for Disease Control and Prevention (2001). "Recommendations for Using Fluoride to Prevent and Control Dental Caries in the United States". MMWR Recommendations and Reports. 50 (RR–14): 1–42. PMID 11521913. Retrieved 14 October 2013.
- Centers for Disease for Control and Prevention (10 July 2013). "Community Water Fluoridation". Retrieved 25 October 2013.
- Chambers, C.; Holliday, A. K. (1975). Modern Inorganic Chemistry: An Intermediate Text (PDF). London: Butterworth & Co. ISBN 978-0-408-70663-6. Archived from the original (PDF) on 23 March 2013.
- Chang, Raymond; Goldsby, Kenneth A. (2013). Chemistry (11th ed.). New York: McGraw-Hill. ISBN 978-0-07-131787-0.
- Cheng, H.; Fowler, D. E.; Henderson, P. B.; Hobbs, J. P.; Pascolini, M. R. (1999). "On the Magnetic Susceptibility of Fluorine". The Journal of Physical Chemistry A. 103 (15): 2861–2866. Bibcode:1999JPCA..103.2861C. doi:10.1021/jp9844720.
- Cheng, K. K.; Chalmers, I.; Sheldon, T. A. (2007). "Adding Fluoride to Water Supplies" (PDF). BMJ. 335 (7622): 699–702. doi:10.1136/bmj.39318.562951.BE. PMC 2001050. PMID 17916854.
- Chisté, V.; Bé, M. M. (2011). "F-18" (PDF). In Bé, M. M.; Coursol, N.; Duchemin, B.; Lagoutine, F.; et al. (eds.). Table de radionucléides (Report). CEA (Commissariat à l'énergie atomique et aux énergies alternatives), LIST, LNE-LNHB (Laboratoire National Henri Becquerel/Commissariat à l'Energie Atomique). Retrieved 15 June 2011.
- Christe, Karl O. (1986). "Chemical Synthesis of Elemental Fluorine". Inorganic Chemistry. 25 (21): 3721–3722. doi:10.1021/ic00241a001.
- Christe Research Group (n.d.). "Chemical Synthesis of Elemental Fluorine". Archived from the original on 4 March 2016. Retrieved 12 January 2013.
- Clark, Jim (2002). "The Acidity of the Hydrogen Halides". chemguide.co.uk. Retrieved 15 October 2013.
- Clayton, Donald (2003). Handbook of Isotopes in the Cosmos: Hydrogen to Gallium. New York: Cambridge University Press. ISBN 978-0-521-82381-4.
- Compressed Gas Association (1999). Handbook of Compressed Gases (4th ed.). Boston: Kluwer Academic Publishers. ISBN 978-0-412-78230-5.
- Cordero, B.; Gómez, V.; Platero-Prats, A. E.; Revés, M.; Echeverría, J.; Cremades, E.; Barragán, F.; Alvarez, S. (2008). "Covalent Radii Revisited". Dalton Transactions (21): 2832–2838. doi:10.1039/b801115j. PMID 18478144.
- Cracher, Connie M. (2012). "Current Concepts in Preventive Dentistry" (PDF). dentalcare.com. Archived from the original (PDF) on 14 October 2013. Retrieved 14 October 2013.
- Croswell, Ken (September 2003). "Fluorine: An element–ary Mystery". Sky and Telescope. Retrieved 17 October 2013.
- Mitchell Crow, James (2011). "First signs of ozone-hole recovery spotted". Nature. doi:10.1038/news.2011.293.
- Davis, Nicole. "Better than blood". Popular Science (November 2006). Archived from the original on 4 June 2011. Retrieved 20 October 2013.
- Davy, Humphry (1813). "Some experiments and observations on the substances produced in different chemical processes on fluor spar". Philosophical Transactions of the Royal Society. 103: 263–279. doi:10.1098/rstl.1813.0034.
- Dean, John A. (1999). Lange's Handbook of Chemistry (15th ed.). New York: McGraw-Hill. ISBN 0-07-016190-9.
- DeBergalis, Michael (2004). "Fluoropolymer films in the photovoltaic industry". Journal of Fluorine Chemistry. 125 (8): 1255–1257. doi:10.1016/j.jfluchem.2004.05.013.
- Directorate-General for the Environment (European Commission) (2007). Trifluralin (PDF) (Report). European Commission. Retrieved 14 October 2013.
- Drews, T.; Supeł, J.; Hagenbach, A.; Seppelt, K. (2006). "Solid State Molecular Structures of Transition Metal Hexafluorides". Inorganic Chemistry. 45 (9): 3782–3788. doi:10.1021/ic052029f. PMID 16634614.
- DuPont (2013a). "Freon". Retrieved 17 October 2013.
- DuPont (2013b). "Understanding the Refrigerant 'R' Nomenclature". Retrieved 17 October 2013.
- Eaton, Charles (1997). "Figure hfl". E-Hand.com: The Electronic Textbook of Hand Surgery. The Hand Center (former practice of Dr. Eaton). Retrieved 28 September 2013.
- Edwards, Philip Neil (1994). "Use of Fluorine in Chemotherapy". In Banks, R. E.; Smart, B. E.; Tatlow, J. C. (eds.). Organofluorine Chemistry: Principles and Commercial Applications. New York: Plenum Press. pp. 501–542. ISBN 978-0-306-44610-8.
- Einstein, F. W. B.; Rao, P. R.; Trotter, J.; Bartlett, N. (1967). "The Crystal Structure of Gold Trifluoride". Journal of the Chemical Society A: Inorganic, Physical, Theoretical. 4: 478–482. doi:10.1039/J19670000478.
- Eisler, Ronald (1995). Sodium Monofluoroacetate (1080) Hazards to Fish, Wildlife and Invertebrates: A Synoptic Review (PDF) (Report). Patuxent Environmental Science Center (U.S. National Biological Service). Retrieved 5 June 2011.
- Ellis, Brian (2001). Scientific Essentialism. Cambridge: Cambridge University Press. ISBN 978-0-521-80094-5.
- El-Kareh, Badih (1994). Fundamentals of Semiconductor Processing Technology. Norwell and Dordrecht: Kluwer Academic Publishers. ISBN 978-0-7923-9534-8.
- El Saadi, M. S.; Hall, A. H.; Hall, P. K.; Riggs, B. S.; Augenstein, W. L.; Rumack, B. H. (1989). "Hydrofluoric Acid Dermal Exposure". Veterinary and Human Toxicology. 31 (3): 243–247. PMID 2741315.
- Emeléus, H. J.; Sharpe, A. G. (1974). Advances in Inorganic Chemistry and Radiochemistry. 16. New York: Academic Press. ISBN 978-0-08-057865-1.
- Emeléus, H. J.; Sharpe, A. G. (1983). Advances in Inorganic Chemistry and Radiochemistry. 27. Academic Press. ISBN 0-12-023627-3.
- Emsley, John (1981). "The Hidden Strength of Hydrogen". New Scientist. 91 (1264): 291–292.
- Emsley, John (2011). Nature's Building Blocks: An A–Z Guide to the Elements (2nd ed.). Oxford: Oxford University Press. ISBN 978-0-19-960563-7.
- Energetics, Inc. (1997). Energy and Environmental Profile of the U.S. Aluminum Industry (PDF) (Report). Retrieved 15 October 2013.
- Filler, R.; Saha, R. (2009). "Fluorine in Medicinal Chemistry: A Century of Progress and a 60-year Retrospective of Selected Highlights" (PDF). Future Medicinal Chemistry. 1 (5): 777–791. doi:10.4155/fmc.09.65. PMID 21426080. Archived from the original (PDF) on 22 October 2013.
- Fischman, Michael L. (2001). "Semiconductor Manufacturing Hazards". In Sullivan, John B.; Krieger, Gary R. (eds.). Clinical Environmental Health and Toxic Exposures (2nd ed.). Philadelphia: Lippincott Williams & Wilkins. pp. 431–465. ISBN 978-0-683-08027-8.
- Forster, P.; Ramaswamy, V.; Artaxo, P.; Berntsen, T.; Betts, R.; Fahey, D. W.; Haywood, J.; Lean, J.; Lowe, D. C.; Myhre, G.; Nganga, J.; Prinn, R.; Raga, G.; Schulz, M.; Van Dorland, R. (2007). "Changes in Atmospheric Constituents and in Radiative Forcing". In Solomon, S.; Manning, M.; Chen, Z.; Marquis, M.; Averyt, K. B.; Tignor, M.; Miller, H. L. (eds.). Climate Change 2007: The Physical Science Basis. Contribution of Working Group I to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change. Cambridge: Cambridge University. pp. 129–234. ISBN 978-0-521-70596-7.
- Fulton, Robert B.; Miller, M. Michael (2006). "Fluorspar". In Kogel, Jessica Elzea; Trivedi, Nikhil C.; Barker, James M.; Krukowski, Stanley T. (eds.). Industrial Minerals & Rocks: Commodities, Markets, and Uses. Littleton: Society for Mining, Metallurgy, and Exploration (U.S.). pp. 461–473. ISBN 978-0-87335-233-8.
- Gabriel, J. L.; Miller Jr, T. F.; Wolfson, M. R.; Shaffer, T. H. (1996). "Quantitative Structure-Activity Relationships of Perfluorinated Hetero-Hydrocarbons as Potential Respiratory Media". ASAIO Journal. 42 (6): 968–973. doi:10.1097/00002480-199642060-00009. PMID 8959271. S2CID 31161098.
- Gains, Paul (18 October 1998). "A New Threat in Blood Doping". The New York Times. Retrieved 18 October 2013.
- Gessner, B. D.; Beller, M.; Middaugh, J. P.; Whitford, G. M. (1994). "Acute Fluoride Poisoning from a Public Water System". New England Journal of Medicine. 330 (2): 95–99. doi:10.1056/NEJM199401133300203. PMID 8259189.
- Giesy, J. P.; Kannan, K. (2002). "Perfluorochemical Surfactants in the Environment". Environmental Science & Technology. 36 (7): 146A–152A. Bibcode:2002EnST...36..146G. doi:10.1021/es022253t. PMID 11999053.
- Godfrey, S. M.; McAuliffe, C. A.; Mackie, A. G.; Pritchard, R. G. (1998). "Inorganic Derivatives of the Elements". In Norman, Nicholas C. (ed.). Chemistry of Arsenic, Antimony and Bismuth. London: Blackie Academic & Professional. pp. 67–158. ISBN 978-0-7514-0389-3.
- Green, S. W.; Slinn, D. S. L.; Simpson, R. N. F.; Woytek, A. J. (1994). "Perfluorocarbon Fluids". In Banks, R. E.; Smart, B. E.; Tatlow, J. C. (eds.). Organofluorine Chemistry: Principles and Applications. New York: Plenum Press. pp. 89–119. ISBN 978-0-306-44610-8.
- Greenwood, N. N.; Earnshaw, A. (1998). Chemistry of the Elements (2nd ed.). Oxford: Butterworth Heinemann. ISBN 0-7506-3365-4.
- Gribble, G. W. (2002). "Naturally Occurring Organofluorines". In Neison, A. H. (ed.). Organofluorines. The Handbook of Environmental Chemistry. 3N. Berlin: Springer. pp. 121–136. doi:10.1007/10721878_5. ISBN 3-540-42064-9.
- Grot, Walter (2011). Fluorinated Ionomers (2nd ed.). Oxford and Waltham: Elsevier. ISBN 978-1-4377-4457-6.
- Hagmann, W. K. (2008). "The Many Roles for Fluorine in Medicinal Chemistry". Journal of Medicinal Chemistry. 51 (15): 4359–4369. doi:10.1021/jm800219f. PMID 18570365.
- Harbison, G. S. (2002). "The Electric Dipole Polarity of the Ground and Low-lying Metastable Excited States of NF". Journal of the American Chemical Society. 124 (3): 366–367. doi:10.1021/ja0159261. PMID 11792193.
- Hasegawa, Y.; Otani, R.; Yonezawa, S.; Takashima, M. (2007). "Reaction Between Carbon Dioxide and Elementary Fluorine". Journal of Fluorine Chemistry. 128 (1): 17–28. doi:10.1016/j.jfluchem.2006.09.002. hdl:10098/1665.
- Haxel, G. B.; Hedrick, J. B.; Orris, G. J. (2005). Stauffer, P. H.; Hendley II, J. W. (eds.). Rare Earth Elements—Critical Resources for High Technology, Fact Sheet 087-02 (Report). U.S. Geological Survey. Retrieved 31 January 2014.
- Haynes, William M., ed. (2011). Handbook of Chemistry and Physics (92nd ed.). Boca Raton: CRC Press. ISBN 978-1-4398-5511-9.
- Hoffman, Robert; Nelson, Lewis; Howland, Mary; Lewin, Neal; Flomenbaum, Neal; Goldfrank, Lewis (2007). Goldfrank's Manual of Toxicologic Emergencies. New York: McGraw-Hill Professional. ISBN 978-0-07-144310-4.
- Honeywell (2006). Recommended medical treatment for hydrofluoric acid exposure (PDF). Morristown: Honeywell International. Archived from the original (PDF) on 8 October 2013. Retrieved 9 January 2014.
- Hoogers, G. (2002). "Fuel Cell Components and Their Impact on Performance". In Hoogers, G. (ed.). Fuel Cell Technology Handbook. Boca Raton: CRC Press. pp. 4–1–4–27. ISBN 0-8493-0877-1.
- Hounshell, David A.; Smith, John Kelly (1988). Science and Corporate Strategy: DuPont R & D, 1902–1980. Cambridge: Cambridge University Press. ISBN 0-521-32767-9.
- Hultén, P.; Höjer, J.; Ludwigs, U.; Janson, A. (2004). "Hexafluorine vs. Standard Decontamination to Reduce Systemic Toxicity After Dermal Exposure to Hydrofluoric Acid". Clinical Toxicology. 42 (4): 355–361. doi:10.1081/CLT-120039541. PMID 15461243. S2CID 27090208.
- ICIS (2 October 2006). "Fluorine's Treasure Trove". Reed Business Information. Retrieved 24 October 2013.
- Jaccaud, M.; Faron, R.; Devilliers, D.; Romano, R. (2000). "Fluorine". Ullmann's Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH. pp. 381–395. doi:10.1002/14356007.a11_293.
- Johnson, Linda A. (28 December 2011). "Against Odds, Lipitor Became World's Top Seller". The Boston Globe. Retrieved 24 October 2013.
- Kacmarek, Robert M.; Wiedemann, Herbert P.; Lavin, Philip T.; Wedel, Mark K.; Tütüncü, Ahmet S.; Slutsky, Arthur S. (2006). "Partial Liquid Ventilation in Adult Patients with Acute Respiratory Distress Syndrome". American Journal of Respiratory and Critical Care Medicine. 173 (8): 882–9. doi:10.1164/rccm.200508-1196OC. PMID 16254269.
- Katakuse, Itsuo; Ichihara, Toshio; Ito, Hiroyuki; Sakurai, Tohru; Matsuo, Takekiyo (1999). "SIMS Experiment". In Arai, T.; Mihama, K.; Yamamoto, K.; Sugano, S. (eds.). Mesoscopic Materials and Clusters: Their Physical and Chemical Properties. Tokyo: Kodansha. pp. 259–273. ISBN 4-06-208635-2.
- Kelly, T. D.; Miller, M. M. (2005). "Historical Fluorspar Statistics". U.S. Geological Service. Retrieved 10 February 2014.
- Keplinger, M. L.; Suissa, L. W. (1968). "Toxicity of Fluorine Short-Term Inhalation". American Industrial Hygiene Association Journal. 29 (1): 10–18. doi:10.1080/00028896809342975. PMID 5667185.
- Kern, S.; Hayward, J.; Roberts, S.; Richardson, J. W.; Rotella, F. J.; Soderholm, L.; Cort, B.; Tinkle, M.; West, M.; Hoisington, D.; Lander, G. A. (1994). "Temperature Variation of the Structural Parameters in Actinide Tetrafluorides". The Journal of Chemical Physics. 101 (11): 9333–9337. Bibcode:1994JChPh.101.9333K. doi:10.1063/1.467963.<
- Khriachtchev, L.; Pettersson, M.; Runeberg, N.; Lundell, J.; Räsänen, M. (2000). "A Stable Argon Compound". Nature. 406 (6798): 874–876. Bibcode:2000Natur.406..874K. doi:10.1038/35022551. PMID 10972285. S2CID 4382128.
- King, D. E.; Malone, R.; Lilley, S. H. (2000). "New Classification and Update on the Quinolone Antibiotics". American Family Physician. 61 (9): 2741–2748. PMID 10821154. Retrieved 8 October 2013.
- Kirsch, Peer (2004). Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications. Weinheim: Wiley-VCH. ISBN 978-3-527-30691-6.
- Kissa, Erik (2001). Fluorinated Surfactants and Repellents (2nd ed.). New York: Marcel Dekker. ISBN 978-0-8247-0472-8.
- Kuriakose, A. K.; Margrave, J. L. (1965). "Kinetics of the Reactions of Elemental Fluorine. IV. Fluorination of Graphite". Journal of Physical Chemistry. 69 (8): 2772–2775. doi:10.1021/j100892a049.
- Lagow, R. J. (1970). The Reactions of Elemental Fluorine; A New Approach to Fluorine Chemistry (PDF) (PhD thesis, Rice University, TX). Ann Arbor: UMI.
- Lau, C.; Anitole, K.; Hodes, C.; Lai, D.; Pfahles-Hutchens, A.; Seed, J. (2007). "Perfluoroalkyl Acids: A Review of Monitoring and Toxicological Findings" (PDF). Toxicological Sciences. 99 (2): 366–394. doi:10.1093/toxsci/kfm128. PMID 17519394.
- Lewars, Errol G. (2008). Modeling Marvels: Computational Anticipation of Novel Molecules. Dordrecht: Springer. ISBN 978-1-4020-6972-7.
- Lide, David R. (2004). Handbook of Chemistry and Physics (84th ed.). Boca Raton: CRC Press. ISBN 0-8493-0566-7.
- Lidin, R.; Molochko, V. A.; Andreeva, L. L. (2000). Химические свойства неорганических веществ [Chemical Properties of Inorganic Substances] (in Russian). Moscow: Khimiya. ISBN 5-7245-1163-0.
- Liteplo, R.; Gomes, R.; Howe, P.; Malcolm, H. (2002). Environmental Health Criteria 227 (Fluoride). Geneva: United Nations Environment Programme; International Labour Organization; World Health Organization. ISBN 92-4-157227-2. Retrieved 14 October 2013.
- Lusty, P. A. J.; Brown, T. J.; Ward, J.; Bloomfield, S. (2008). "The Need for Indigenous Fluorspar Production in England". British Geological Survey. Retrieved 13 October 2013.
- Mackay, Kenneth Malcolm; Mackay, Rosemary Ann; Henderson, W. (2002). Introduction to Modern Inorganic Chemistry (6th ed.). Cheltenham: Nelson Thornes. ISBN 0-7487-6420-8.
- Macomber, Roger (1996). Organic chemistry. 1. Sausalito: University Science Books. ISBN 978-0-935702-90-3.
- Marggraf, Andreas Sigismun (1770). "Observation concernant une volatilisation remarquable d'une partie de l'espece de pierre, à laquelle on donne les noms de flosse, flüsse, flus-spaht, et aussi celui d'hesperos; laquelle volatilisation a été effectuée au moyen des acides" [Observation of a remarkable volatilization of part of a type of stone to which one gives the name flosse, flüsse, flus-spaht, as well as that of hesperos; which volatilization was effected by means of acids]. Mémoires de l'Académie royale des sciences et belles-lettres (in French). XXIV: 3–11.
- Martin, John W., ed. (2007). Concise Encyclopedia of the Structure of Materials. Oxford and Amsterdam: Elsevier. ISBN 978-0-08-045127-5.
- Marya, C. M. (2011). A Textbook of Public Health Dentistry. New Delhi: Jaypee Brothers Medical Publishers. ISBN 978-93-5025-216-1.
- Matsui, M. (2006). "Fluorine-containing Dyes". In Kim, Sung-Hoon (ed.). Functional dyes. Orlando: Academic Press. pp. 257–266. ISBN 978-0-12-412490-5.
- Meusinger, Reinhard; Chippendale, A. Margaret; Fairhurst, Shirley A. (2012). "Nuclear Magnetic Resonance and Electron Spin Resonance Spectroscopy". Ullmann's Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH. pp. 609–660. doi:10.1002/14356007.b05_471.
- Meyer, Eugene (1977). Chemistry of Hazardous Materials. Englewood Cliffs: Prentice Hall. ISBN 978-0-13-129239-0.
- Miller, M. Michael (2003a). "Fluorspar" (PDF). U.S. Geological Survey Minerals Yearbook. U.S. Geological Survey. pp. 27.1–27.12.
- Miller, M. Michael (2003b). "Mineral Resource of the Month, Fluorspar" (PDF). U.S. Geological Survey. Retrieved 24 October 2013.
- Mitchell, E. Siobhan (2004). Antidepressants. New York: Chelsea House Publishers. ISBN 978-1-4381-0192-7.
- Moeller, T.; Bailar, J. C.; Kleinberg (1980). Chemistry, with Inorganic Qualitative Analysis (3rd ed.). New York: Academic Press. ISBN 0-12-503350-8.
- Moissan, Henri (1886). "Action d'un courant électrique sur l'acide fluorhydrique anhydre". Comptes rendus hebdomadaires des séances de l'Académie des sciences (in French). 102: 1543–1544. Retrieved 9 October 2013.
- McCoy, M. (2007). "SURVEY Market Challenges Dim the Confidence of the World's Chemical CEOs". Chemical & Engineering News. 85 (23): 11. doi:10.1021/cen-v085n023.p011a.
- Moore, John W.; Stanitski, Conrad L.; Jurs, Peter C. (2010). Principles of Chemistry: The Molecular Science. Belmont: Brooks/Cole. ISBN 978-0-495-39079-4.
- Morrow, S. I.; Perry, D. D.; Cohen, M. S. (1959). "The Formation of Dinitrogen Tetrafluoride in the Reaction of Fluorine and Ammonia". Journal of the American Chemical Society. 81 (23): 6338–6339. doi:10.1021/ja01532a066.
- Müller, Peter (2009). "5.067 Crystal Structure Refinement" (PDF). Cambridge: MIT OpenCourseWare. Retrieved 13 October 2013.
- Murphy, C. D.; Schaffrath, C.; O'Hagan, D. (2003). "Fluorinated Natural Products: The Biosynthesis of Fluoroacetate and 4-Fluorothreonine in Streptomyces cattleya". Chemosphere. 52 (2): 455–461. Bibcode:2003Chmsp..52..455M. doi:10.1016/S0045-6535(03)00191-7. PMID 12738270.
- Murthy, C. Parameshwara; Mehdi Ali, S. F.; Ashok, D. (1995). University Chemistry. I. New Delhi: New Age International. ISBN 978-81-224-0742-6.
- National Health and Medical Research Council (2007). A Systematic Review of the Efficacy and Safety of Fluoridation, Part A: Review of Methodology and Results (PDF). Canberra: Australian Government. ISBN 1-86496-421-9. Archived from the original (PDF) on 13 January 2012. Retrieved 8 October 2013.
- The National Institute for Occupational Safety and Health (1994). "Fluorine". Documentation for Immediately Dangerous To Life or Health Concentrations (IDLHs). Retrieved 15 January 2014.
- The National Institute for Occupational Safety and Health (1994). "Chlorine". Documentation for Immediately Dangerous To Life or Health Concentrations (IDLHs). Retrieved 13 July 2014.
- National Nuclear Data Center. "NuDat 2.1 Database". Brookhaven National Laboratory. Retrieved 25 October 2013.
- National Oceanic and Atmospheric Administration. "UN/NA 1045 (United Nations/North America Fluorine Data Sheet)". Retrieved 15 October 2013.
- Navarrini, Walter; Venturini, Francesco; Tortelli, Vito; Basak, Soubir; Pimparkar, Ketan P.; Adamo, Andrea; Jensen, Klavs F. (2012). "Direct fluorination of carbon monoxide in microreactors". Journal of Fluorine Chemistry. 142: 19–23. doi:10.1016/j.jfluchem.2012.06.006.
- Nelson, Eugene W. (1947). "'Bad Man' of The Elements". Popular Mechanics. 88 (2): 106–108, 260.
- Nelson, J. M.; Chiller, T. M.; Powers, J. H.; Angulo, F. J. (2007). "Food Safety: Fluoroquinolone‐ResistantCampylobacterSpecies and the Withdrawal of Fluoroquinolones from Use in Poultry: A Public Health Success Story" (PDF). Clinical Infectious Diseases. 44 (7): 977–980. doi:10.1086/512369. PMID 17342653.
- Nielsen, Forrest H. (2009). "Micronutrients in Parenteral Nutrition: Boron, Silicon, and Fluoride". Gastroenterology. 137 (5): S55–60. doi:10.1053/j.gastro.2009.07.072. PMID 19874950. Retrieved 29 April 2018.
- Norwood, Charles J.; Fohs, F. Julius (1907). Kentucky Geological Survey, Bulletin No. 9: Fluorspar Deposits of Kentucky. Kentucky Geological Survey.
- Noury, S.; Silvi, B.; Gillespie, R. J. (2002). "Chemical Bonding in Hypervalent Molecules: Is the Octet Rule Relevant?" (PDF). Inorganic Chemistry. 41 (8): 2164–2172. doi:10.1021/ic011003v. PMID 11952370. Retrieved 23 May 2012.
- O'Hagan, D. (2008). "Understanding Organofluorine Chemistry. An Introduction to the C–F Bond". Chemical Society Reviews. 37 (2): 308–319. doi:10.1039/b711844a. PMID 18197347.
- O'Hagan, D.; Schaffrath, C.; Cobb, S. L.; Hamilton, J. T. G.; Murphy, C. D. (2002). "Biochemistry: Biosynthesis of an Organofluorine Molecule". Nature. 416 (6878): 279. Bibcode:2002Natur.416..279O. doi:10.1038/416279a. PMID 11907567. S2CID 4415511.
- Okada, T.; Xie, G.; Gorseth, O.; Kjelstrup, S.; Nakamura, N.; Arimura, T. (1998). "Ion and Water Transport Characteristics of Nafion Membranes as Electrolytes". Electrochimica Acta. 43 (24): 3741–3747. doi:10.1016/S0013-4686(98)00132-7.
- Okazoe, T. (2009). "Overview on the History of Organofluorine Chemistry from the Viewpoint of Material Industry". Proceedings of the Japan Academy, Series B. 85 (8): 276–289. Bibcode:2009PJAB...85..276O. doi:10.2183/pjab.85.276. PMC 3621566. PMID 19838009.
- Olivares, M.; Uauy, R. (2004). Essential Nutrients in Drinking Water (Draft) (PDF) (Report). World Health Organization (WHO). Archived from the original (PDF) on 19 October 2012. Retrieved 14 October 2013.
- Parente, Luca (2001). "The Development of Synthetic Glucocorticoids". In Goulding, Nicolas J.; Flower, Rod J. (eds.). Glucocorticoids. Basel: Birkhäuser. pp. 35–53. ISBN 978-3-7643-6059-7.
- Partington, J. R. (1923). "The early history of hydrofluoric acid". Memoirs and Proceedings of the Manchester Literary and Philosophical Society. 67 (6): 73–87.
- Patnaik, Pradyot (2007). A Comprehensive Guide to the Hazardous Properties of Chemical Substances (3rd ed.). Hoboken: John Wiley & Sons. ISBN 978-0-471-71458-3.
- Pauling, Linus (1960). The Nature of the Chemical Bond (3rd ed.). Ithaca: Cornell University Press. ISBN 978-0-8014-0333-0.
- Pauling, L.; Keaveny, I.; Robinson, A. B. (1970). "The Crystal Structure of α-Fluorine". Journal of Solid State Chemistry. 2 (2): 225–227. Bibcode:1970JSSCh...2..225P. doi:10.1016/0022-4596(70)90074-5.
- Perry, Dale L. (2011). Handbook of Inorganic Compounds (2nd ed.). Boca Raton: CRC Press. ISBN 978-1-4398-1461-1.
- Pitzer, K. S. (1975). "Fluorides of Radon and Element 118". Journal of the Chemical Society, Chemical Communications (18): 760b–761. doi:10.1039/C3975000760B.
- Pitzer, Kenneth S., ed. (1993). Molecular Structure and Statistical Thermodynamics: Selected Papers of Kenneth S. Pitzer. Singapore: World Scientific Publishing. ISBN 978-981-02-1439-5.
- Pizzo, G.; Piscopo, M. R.; Pizzo, I.; Giuliana, G. (2007). "Community Water Fluoridation and Caries Prevention: A Critical Review" (PDF). Clinical Oral Investigations. 11 (3): 189–193. doi:10.1007/s00784-007-0111-6. PMID 17333303. S2CID 13189520.
- Posner, Stefan (2011). "Perfluorinated Compounds: Occurrence and Uses in Products". In Knepper, Thomas P.; Large, Frank T. (eds.). Polyfluorinated Chemicals and Transformation Products. Heidelberg: Springer Science+Business Media. pp. 25–40. ISBN 978-3-642-21871-2.
- Posner, Stefan; et al. (2013). Per- and Polyfluorinated Substances in the Nordic Countries: Use Occurrence and Toxicology. Copenhagen: Nordic Council of Ministers. doi:10.6027/TN2013-542. ISBN 978-92-893-2562-2.
- Preskorn, Sheldon H. (1996). Clinical Pharmacology of Selective Serotonin Reuptake Inhibitors. Caddo: Professional Communications. ISBN 978-1-884735-08-0.
- Principe, Lawrence M. (2012). The Secrets of Alchemy. Chicago: University of Chicago Press. ISBN 978-0-226-68295-2.
- Proudfoot, A. T.; Bradberry, S. M.; Vale, J. A. (2006). "Sodium Fluoroacetate Poisoning". Toxicological Reviews. 25 (4): 213–219. doi:10.2165/00139709-200625040-00002. PMID 17288493. S2CID 29189551.
- PRWeb (28 October 2010). "Global Fluorochemicals Market to Exceed 2.6 Million Tons by 2015, According to a New Report by Global Industry Analysts, Inc". prweb.com. Retrieved 24 October 2013.
- PRWeb (23 February 2012). "Global Fluorspar Market to Reach 5.94 Million Metric Tons by 2017, According to New Report by Global Industry Analysts, Inc". prweb.com. Retrieved 24 October 2013.
- PRWeb (7 April 2013). "Fluoropolymers Market is Poised to Grow at a CAGR of 6.5% & to Reach $9,446.0 Million by 2016 – New report by MarketsandMarkets". prweb.com. Retrieved 24 October 2013.
- Pyykkö, Pekka; Atsumi, Michiko (2009). "Molecular Double-Bond Covalent Radii for Elements Li–E112". Chemistry: A European Journal. 15 (46): 12770–9. doi:10.1002/chem.200901472. PMID 19856342.
- Raghavan, P. S. (1998). Concepts and Problems in Inorganic Chemistry. Delhi: Discovery Publishing House. ISBN 978-81-7141-418-5.
- Raj, P. Prithvi; Erdine, Serdar (2012). Pain-Relieving Procedures: The Illustrated Guide. Chichester: John Wiley & Sons. ISBN 978-0-470-67038-5.
- Ramkumar, Jayshree (2012). "Nafion Perfluorosulphonate Membrane: Unique Properties and Various Applications". In Banerjee, S.; Tyagi, A. K. (eds.). Functional Materials: Preparation, Processing and Applications. London and Waltham: Elsevier. pp. 549–578. ISBN 978-0-12-385142-0.
- Reddy, D. (2009). "Neurology of Endemic Skeletal Fluorosis". Neurology India. 57 (1): 7–12. doi:10.4103/0028-3886.48793. PMID 19305069.
- Renda, Agostino; Fenner, Yeshe; Gibson, Brad K.; Karakas, Amanda I.; Lattanzio, John C.; Campbell, Simon; Chieffi, Alessandro; Cunha, Katia; Smith, Verne V. (2004). "On the origin of fluorine in the Milky Way". Monthly Notices of the Royal Astronomical Society. 354 (2): 575–580. arXiv:astro-ph/0410580. Bibcode:2004MNRAS.354..575R. doi:10.1111/j.1365-2966.2004.08215.x. S2CID 12330666.
- Renner, R. (2006). "The Long and the Short of Perfluorinated Replacements". Environmental Science & Technology. 40 (1): 12–13. Bibcode:2006EnST...40...12R. doi:10.1021/es062612a. PMID 16433328.
- Rhoades, David Walter (2008). Broadband Dielectric Spectroscopy Studies of Nafion (PhD dissertation, University of Southern Mississippi, MS). Ann Arbor: ProQuest. ISBN 978-0-549-78540-8.
- Richter, M.; Hahn, O.; Fuchs, R. (2001). "Purple Fluorite: A Little Known Artists' Pigment and Its Use in Late Gothic and Early Renaissance Painting in Northern Europe". Studies in Conservation. 46 (1): 1–13. doi:10.1179/sic.2001.46.1.1. JSTOR 1506878. S2CID 191611885.
- Riedel, Sebastian; Kaupp, Martin (2009). "The highest oxidation states of the transition metal elements". Coordination Chemistry Reviews. 253 (5–6): 606–624. doi:10.1016/j.ccr.2008.07.014.
- Ripa, L. W. (1993). "A Half-century of Community Water Fluoridation in the United States: Review and Commentary" (PDF). Journal of Public Health Dentistry. 53 (1): 17–44. doi:10.1111/j.1752-7325.1993.tb02666.x. PMID 8474047. Archived from the original (PDF) on 4 March 2009.
- Roblin, I.; Urban, M.; Flicoteau, D.; Martin, C.; Pradeau, D. (2006). "Topical Treatment of Experimental Hydrofluoric Acid Skin Burns by 2.5% Calcium Gluconate". Journal of Burn Care & Research. 27 (6): 889–894. doi:10.1097/01.BCR.0000245767.54278.09. PMID 17091088. S2CID 3691306.
- Salager, Jean-Louis (2002). Surfactants: Types and Uses (PDF). FIRP Booklet # 300-A. Laboratory of Formulation, Interfaces, Rheology, and Processes, Universidad de los Andes. Retrieved 13 October 2013.
- Sandford, Graham (2000). "Organofluorine Chemistry". Philosophical Transactions. 358 (1766): 455–471. Bibcode:2000RSPTA.358..455S. doi:10.1098/rsta.2000.0541. S2CID 202574641.
- Sarkar, S. (2008). "Artificial Blood". Indian Journal of Critical Care Medicine. 12 (3): 140–144. doi:10.4103/0972-5229.43685. PMC 2738310. PMID 19742251.
- Scheele, Carl Wilhelm (1771). "Undersŏkning om fluss-spat och dess syra" [Investigation of Fluorite and Its Acid]. Kungliga Svenska Vetenskapsademiens Handlingar [Proceedings of the Royal Swedish Academy of Science] (in Swedish). 32: 129–138.
- Schimmeyer, S. (2002). "The Search for a Blood Substitute". Illumin. Columbia: University of Southern Carolina. 15 (1). Archived from the original on 2 October 2011. Retrieved 15 October 2013.
- Schlöder, T.; Riedel, S. (2012). "Investigation of Heterodimeric and Homodimeric Radical Cations of the Series: [F2O2]+, [F2Cl2]+, [Cl2O2]+, [F4]+, and [Cl4]+". RSC Advances. Royal Society of Chemistry. 2 (3): 876–881. doi:10.1039/C1RA00804H.
- Schmedt Auf Der Günne, Jörn; Mangstl, Martin; Kraus, Florian (2012). "Occurrence of Difluorine F2in Nature-In Situ Proof and Quantification by NMR Spectroscopy". Angewandte Chemie International Edition. 51 (31): 7847–9. doi:10.1002/anie.201203515. PMID 22763992.
- Schmitz, A.; Kälicke, T.; Willkomm, P.; Grünwald, F.; Kandyba, J.; Schmitt, O. (2000). "Use of Fluorine-18 Fluoro-2-deoxy-D-glucose Positron Emission Tomography in Assessing the Process of Tuberculous Spondylitis" (PDF). Journal of Spinal Disorders. 13 (6): 541–544. doi:10.1097/00002517-200012000-00016. PMID 11132989. Retrieved 8 October 2013.
- Schulze-Makuch, D.; Irwin, L. N. (2008). Life in the Universe: Expectations and Constraints (2nd ed.). Berlin: Springer-Verlag. ISBN 978-3-540-76816-6.
- Schwarcz, Joseph A. (2004). The Fly in the Ointment: 70 Fascinating Commentaries on the Science of Everyday Life. Toronto: ECW Press. ISBN 1-55022-621-5.
- Senning, A. (2007). Elsevier's Dictionary of Chemoetymology: The Whies and Whences of Chemical Nomenclature and Terminology. Amsterdam and Oxford: Elsevier. ISBN 978-0-444-52239-9.
- Shaffer, T. H.; Wolfson, M. R.; Clark Jr, L. C. (1992). "Liquid Ventilation". Pediatric Pulmonology. 14 (2): 102–109. doi:10.1002/ppul.1950140208. PMID 1437347.
- Shin, Richard D.; Silverberg, Mark A. (2013). "Fluoride Toxicity". Medscape. Retrieved 15 October 2013.
- Shriver, Duward; Atkins, Peter (2010). Solutions Manual for Inorganic Chemistry. New York: W. H. Freeman. ISBN 978-1-4292-5255-3.
- Shulman, J. D.; Wells, L. M. (1997). "Acute Fluoride Toxicity from Ingesting Home-use Dental Products in Children, Birth to 6 Years of Age". Journal of Public Health Dentistry. 57 (3): 150–158. doi:10.1111/j.1752-7325.1997.tb02966.x. PMID 9383753.
- Siegemund, G. N.; Schwertfeger, W.; Feiring, A.; Smart, B.; Behr, F.; Vogel, H.; McKusick, B. (2000). "Fluorine Compounds, Organic". Ullmann's Encyclopedia of Industrial Chemistry. 15. Weinheim: Wiley-VCH. doi:10.1002/14356007.a11_349.
- Slye, Orville M. (2012). "Fire Extinguishing Agents". In Ullmann, Franz (ed.). Ullmann's Encyclopedia of Industrial Chemistry. 15. Weinheim: Wiley-VCH. pp. 1–11. doi:10.1002/14356007.a11_113.pub2. ISBN 978-3527306732.
- Steenland, K.; Fletcher, T.; Savitz, D. A. (2010). "Epidemiologic Evidence on the Health Effects of Perfluorooctanoic Acid (PFOA)". Environmental Health Perspectives. 118 (8): 1100–1108. doi:10.1289/ehp.0901827. PMC 2920088. PMID 20423814.
- Stillman, John Maxson (December 1912). "Basil Valentine, A Seventeenth Century Hoax". Popular Science Monthly. 81. Retrieved 14 October 2013.
- Storer, Frank H. (1864). First Outlines of a Dictionary of Solubilities of Chemical Substances. Cambridge: Sever and Francis.
- Swinson, Joel (June 2005). "Fluorine – A Vital Element in the Medicine Chest" (PDF). PharmaChem. Pharmaceutical Chemistry: 26–27. Archived from the original (PDF) on 8 February 2012. Retrieved 9 October 2013.
- Taber, Andrew (22 April 1999). "Dying to ride". Salon. Retrieved 18 October 2013.
- Tanner Industries (January 2011). "Anhydrous Ammonia: (MSDS) Material Safety Data Sheet". tannerind.com. Retrieved 24 October 2013.
- Theodoridis, George (2006). "Fluorine-Containing Agrochemicals: An Overview of Recent Developments". In Tressaud, Alain (ed.). Fluorine and the Environment : Agrochemicals, Archaeology, Green Chemistry & Water. Amsterdam and Oxford: Elsevier. pp. 121–176. ISBN 978-0-444-52672-4.
- Toon, Richard (1 September 2011). "The discovery of fluorine". Education in Chemistry. Vol. 48 no. 5. Royal Society of Chemistry. pp. 148–151. ISSN 0013-1350.
- Transparency Market Research (17 May 2013). "Fluorochemicals Market is Expected to Reach USD 21.5 Billion Globally by 2018: Transparency Market Research". Transparency Market Research Blog. Archived from the original on 22 February 2014. Retrieved 15 October 2013.
- Ullmann, Fritz (2008). Ullmann's Fibers (2 volumes). Weinheim: Wiley-VCH. ISBN 978-3-527-31772-1.
- United States Environmental Protection Agency (1996). "R.E.D. Facts: Trifluralin" (PDF). Archived from the original (PDF) on 18 October 2013. Retrieved 17 October 2013.
- United States Environmental Protection Agency (2012). "Emerging Contaminants – Perfluorooctane Sulfonate (PFOS) and Perfluorooctanoic Acid (PFOA)" (PDF). Archived from the original (PDF) on 29 October 2013. Retrieved 4 November 2013.
- United States Environmental Protection Agency (2013a). "Class I Ozone-depleting Substances". Archived from the original on 10 December 2010. Retrieved 15 October 2013.
- United States Environmental Protection Agency (2013b). "Phaseout of HCFCs (Class II Ozone-Depleting Substances)". Retrieved 15 October 2013.
- Viel, Claude; Goldwhite, Harold (1993). "1906 Nobel Laureate: Henri Moissan, 1852–1907". In Laylin, K. James (ed.). Nobel Laureates in Chemistry, 1901–1992. Washington: American Chemical Society; Chemical Heritage Foundation. pp. 35–41. ISBN 978-0-8412-2690-6.
- Vigoureux, P. (1961). "The Gyromagnetic Ratio of the Proton". Contemporary Physics. 2 (5): 360–366. Bibcode:1961ConPh...2..360V. doi:10.1080/00107516108205282.
- Villalba, Gara; Ayres, Robert U.; Schroder, Hans (2008). "Accounting for Fluorine: Production, Use, and Loss". Journal of Industrial Ecology. 11: 85–101. doi:10.1162/jiec.2007.1075.
- Walsh, Kenneth A. (2009). Beryllium Chemistry and Processing. Materials Park: ASM International. ISBN 978-0-87170-721-5.
- Walter, P. (2013). "Honeywell Invests $300m in Green Refrigerant". Chemistry World.
- Weeks, M. E. (1932). "The Discovery of the Elements. XVII. The Halogen Family". Journal of Chemical Education. 9 (11): 1915–1939. Bibcode:1932JChEd...9.1915W. doi:10.1021/ed009p1915.
- Werner, N. L.; Hecker, M. T.; Sethi, A. K.; Donskey, C. J. (2011). "Unnecessary use of Fluoroquinolone Antibiotics in Hospitalized Patients". BMC Infectious Diseases. 11: 187–193. doi:10.1186/1471-2334-11-187. PMC 3145580. PMID 21729289.
- Wiberg, Egon; Wiberg, Nils; Holleman, Arnold Frederick (2001). Inorganic Chemistry. San Diego: Academic Press. ISBN 978-0-12-352651-9.
- Willey, Ronald R. (2007). Practical Equipment, Materials, and Processes for Optical Thin Films. Charlevoix: Willey Optical. ISBN 978-0-615-14397-2.
- Yaws, Carl L.; Braker, William (2001). "Fluorine". Matheson Gas Data Book (7th ed.). Parsippany: Matheson Tri-Gas. ISBN 978-0-07-135854-5.
- Yeung, C. A. (2008). "A Systematic Review of the Efficacy and Safety of Fluoridation". Evidence-Based Dentistry. 9 (2): 39–43. doi:10.1038/sj.ebd.6400578. PMID 18584000.
- Young, David A. (1975). Phase Diagrams of the Elements (Report). Lawrence Livermore Laboratory. Retrieved 10 June 2011.
- Zareitalabad, P.; Siemens, J.; Hamer, M.; Amelung, W. (2013). "Perfluorooctanoic acid (PFOA) and perfluorooctanesulfonic acid (PFOS) in surface waters, sediments, soils and wastewater – A review on concentrations and distribution coefficients". Chemosphere. 91 (6): 725–32. Bibcode:2013Chmsp..91..725Z. doi:10.1016/j.chemosphere.2013.02.024. PMID 23498059.
- Zorich, Robert (1991). Handbook of Quality Integrated Circuit Manufacturing. San Diego: Academic Press. ISBN 978-0-323-14055-3.