Congenital distal spinal muscular atrophy
Congenital distal spinal muscular atrophy is a hereditary condition characterized by muscle wasting (atrophy), particularly of distal muscles in legs and hands, and by early-onset contractures (permanent shortening of a muscle or joint) of the hip, knee, and ankle. Affected individuals often have shorter lower limbs relative to the trunk and upper limbs. The condition is a result of a loss of anterior horn cells localized to lumbar and cervical regions of the spinal cord early in infancy, which in turn is caused by a mutation of the TRPV4 gene. The disorder is inherited in an autosomal dominant manner.[1] Arm muscle and function, as well as cardiac and respiratory functions are typically well preserved.[2]
| Congenital distal spinal muscular atrophy | |
|---|---|
| Other names | congenital dSMA |
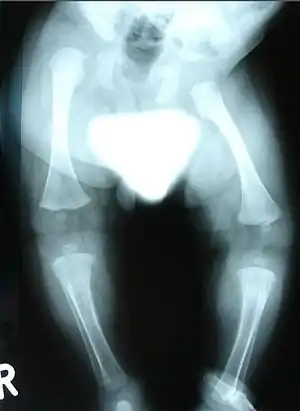 | |
| Radiograph showing dysplasia in lower limbs | |
| Specialty | Pediatrics, medical genetics |
Signs and symptoms
- Neurogenic muscle weakness
- Atrophy (of lower and upper limbs)
- Club foot
- Arthrogryposis
- Scoliosis
- Platyspondyly
- Pes cavus
- Vocal cord paralysis
Causes
Congenital distal spinal muscular atrophy is caused by a mutation of the TRPV4 gene found on the 12q23-12q24.1.[3] The mutation causes an affected individual to have lower levels of TRPV4 expression. This deficiency can lead to abnormal osmotic regulation. Congenital dSMA is genetically heterogeneous, meaning a mutation on this gene can cause a plethora of other phenotypically related or phenotypically unrelated diseases depending on the region that is mutated.
Pathophysiology
The TRPV4 (transient receptor potential vanilloid 4) gene, located on chromosome 12, encodes for a protein that serves as an ion channel, typically found in the plasma membrane and is permeable to Ca2+. Abnormal regulation of Ca2+ can lead to inefficient muscle contraction.[4] TRPV4 plays a major role in mechanosensation, as well as osmosensory functions in nerve endings, endothoelia, and alveoli.[5] The TRPV4 protein consists of 871 amino acids with its N- and C- terminals facing intracellularly. The protein also contains six alpha helices that pass through the plasma membrane. Mutations in TRPV4 can result in the loss of its normal function, or a toxic gain of function. In the latter case, intracellular Ca2+ levels are increased, which results in abnormal regulation.[6]
Mechanism
The ankyrin repeat domain (ARD) is a region located near the intracellular N-terminal of the TRPV4 protein and consists of six ankyrin repeats. Four missense mutations have been identified at three specific positions all located within the ARD. All of these mutations are due to the swapping out of arginine with a different amino acid.[7] Arginine is highly polar and positively charged, while its replacements are less polar or nonpolar. Some of these identified amino acid substitutions are:
- R296H, arginine to histidine substitution
- R315W, arginine to tryptophan substitution
- R316C, arginine to cysteine substitution
- R594H, arginine to histidine substitution
Diagnosis
Electrophysiological evidence of denervation with intact motor and sensory nerve conduction findings must be made by using nerve conduction studies, usually in conjunction with EMG. The presence of polyphasic potentials and fibrillation at rest are characteristic of congenital dSMA.[6] The following are useful in diagnosis:
- Nerve conduction studies (NCS), to test for denervation
- Electromyography (EMG), also to detect denervation
- X-ray, to look for bone abnormalities
- Magnetic resonance imaging (MRI)
- Skeletal muscle biopsy examination
- Serum creatine kinase (CK) level in blood, usually elevated in affected individuals
- Pulmonary function test
Management
Congenital dSMA has a relatively stable disease course, with disability mainly attributed to increased contractures rather than loss of muscle strength. Individuals frequently use crutches, knee, ankle, and/or foot orthoses, or wheelchairs.[2] Orthopaedic surgery can be an option for some patients with severely impaired movement. Physical therapy and occupational therapy can help prevent further contractures from occurring, though they do not reverse the effects of preexisting ones. Some literature suggests the use of electrical stimulation or botulinum toxin to halt the progression of contractures.[8]
See also
References
- Oates EC, Reddel S, Rodriguez ML, et al. (June 2012). "Autosomal dominant congenital spinal muscular atrophy: a true form of spinal muscular atrophy caused by early loss of anterior horn cells". Brain. 135 (Pt 6): 1714–23. doi:10.1093/brain/aws108. PMID 22628388.
- Mercuri E, Messina S, Kinali M, et al. (February 2004). "Congenital form of spinal muscular atrophy predominantly affecting the lower limbs: a clinical and muscle MRI study". Neuromuscul. Disord. 14 (2): 125–9. doi:10.1016/j.nmd.2003.09.005. PMID 14733958.
- Everaerts W, Nilius B, Owsianik G (September 2010). "The vanilloid transient receptor potential channel TRPV4': from structure to disease". Prog. Biophys. Mol. Biol. 103 (1): 2–17. doi:10.1016/j.pbiomolbio.2009.10.002. PMID 19835908.
- Menezes MP, North KN (June 2012). "Inherited neuromuscular disorders: pathway to diagnosis". J Paediatr Child Health. 48 (6): 458–65. doi:10.1111/j.1440-1754.2011.02210.x. PMID 22050238.
- Auer-Grumbach M, Olschewski A, Papić L, et al. (February 2010). "Alterations in the ankyrin domain of TRPV4 cause congenital distal SMA, scapuloperoneal SMA and HMSN2C". Nat. Genet. 42 (2): 160–4. doi:10.1038/ng.508. PMC 3272392. PMID 20037588.
- Fiorillo C, Moro F, Brisca G, et al. (August 2012). "TRPV4 mutations in children with congenital distal spinal muscular atrophy". Neurogenetics. 13 (3): 195–203. doi:10.1007/s10048-012-0328-7. PMID 22526352.
- Dai J, Cho TJ, Unger S, et al. (July 2010). "TRPV4'-pathy, a novel channelopathy affecting diverse systems". J. Hum. Genet. 55 (7): 400–2. doi:10.1038/jhg.2010.37. PMID 20505684.
- Farmer SE, James M (September 2001). "Contractures in orthopaedic and neurological conditions: a review of causes and treatment". Disabil Rehabil. 23 (13): 549–58. doi:10.1080/09638280010029930. PMID 11451189.