Primary lateral sclerosis
Primary lateral sclerosis (PLS) is a very rare neuromuscular disease characterized by progressive muscle weakness in the voluntary muscles. PLS belongs to a group of disorders known as motor neuron diseases. Motor neuron diseases develop when the nerve cells that control voluntary muscle movement degenerate and die, causing weakness in the muscles they control.
| Primary lateral sclerosis | |
|---|---|
| Other names | Adult-onset primary lateral sclerosis [1] |
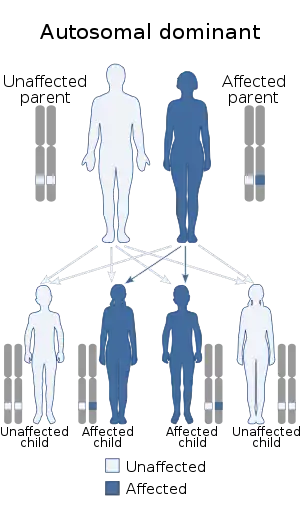 | |
| This condition is inherited via an autosomal dominant manner[2] | |
| Specialty | Neurology |
PLS only affects upper motor neurons.[3] There is no evidence of the degeneration of spinal motor neurons or muscle wasting (amyotrophy) that occurs in amyotrophic lateral sclerosis (ALS).
Symptoms
Onset of PLS usually occurs spontaneously after age 50 and progresses gradually over a number of years, or even decades. The disorder usually begins in the legs, but it may start in the tongue or the hands. Symptoms may include difficulty with balance, weakness and stiffness in the legs, and clumsiness. Other common symptoms are spasticity (involuntary muscle contraction due to the stretching of muscle, which depends on the velocity of the stretch) in the hands, feet, or legs, foot dragging, and speech and swallowing problems due to involvement of the facial muscles. Breathing may also become compromised in the later stages of the disease, causing those patients who develop ventilatory failure to require noninvasive ventilatory support.[4] Hyperreflexia is another key feature of PLS as seen in patients presenting with the Babinski's sign.[5] Some people present with emotional lability and bladder urgency,[5] and occasionally people with PLS experience mild cognitive changes detectable on neuropsychological testing, particularly on measures of executive function.[6]
PLS is not considered hereditary when onset is in adulthood; however, juvenile primary lateral sclerosis (JPLS) has been linked to a mutation in the ALS2 gene which encodes the cell-signalling protein alsin.[7]
The issue of whether PLS exists as a different entity from ALS is not clear, as some patients initially diagnosed as having PLS ultimately develop lower motor neuron signs.[8][9] When this happens it is classed as ALS.[10]
Spasticity
Primary lateral sclerosis (PLS) usually presents with gradual-onset, progressive, lower-extremity stiffness and pain due to muscle spasticity. Onset is often asymmetrical.[4] Although the muscles do not appear to atrophy as in ALS (at least initially), the disabling aspect of PLS is muscle spasticity and cramping, and intense pain when those muscles are stretched, resulting in joint immobility. A normal walking stride may become a tiny step shuffle with related instability and falling.
Cause
Researchers do not fully understand what causes PLS, although it is thought it could be due to a combination of environmental and genetic factors.[11] Studies are being done to evaluate the possible causes, although linking causality can be difficult due to the relatively low number of people who are diagnosed with PLS.
Juvenile PLS may be caused by the ALS2 gene, although this condition is very rare.
Diagnosis
There are no specific tests for the diagnosis of PLS. Therefore, the diagnosis occurs as the result of eliminating other possible causes of the symptoms and by an extended observation period.[12]
Like ALS, diagnosing PLS is a diagnosis of exclusion, as there is no one test that can confirm a diagnosis of PLS. The Pringle Criteria,[13] proposed by Pringle et al, provides a guideline of nine points that, if confirmed, can suggest a diagnosis of PLS. Due to the fact that a person with ALS may initially present with only upper motor neuron symptoms, indicative of PLS, one key aspect of the Pringle Criteria is requiring a minimum of three years between symptom onset and symptom diagnosis. When these criteria are met, a diagnosis of PLS is highly likely.[14] Other aspects of Pringle Criteria include normal EMG findings, thereby ruling out lower motor neuron involvement that is indicative of ALS, and absence of family history for Hereditary Spastic Paraplegia (HSP) and ALS. Imaging studies to rule out structural or demyelinating lesions may be done as well. Hoffman's sign and Babinski reflex may be present and indicative of upper motor neuron damage.
Treatment
Treatment for individuals with PLS is symptomatic. Baclofen and tizanidine may reduce spasticity. Quinine or phenytoin may decrease cramps. Some patients who do not receive adequate relief from oral treatment may consider intrathecal baclofen (i.e., infusion of medication directly into the cerebrospinal fluid via a surgically placed continuous infusion pump). However, patients are carefully selected for this type of procedure to ensure that they will likely benefit from this invasive procedure.[4]
Physical therapy often helps prevent joint immobility. Speech therapy may be useful for those with involvement of the facial muscles. Physiotherapy treatment focuses on reducing muscle tone, maintaining or improving range of motion, increasing strength and coordination, and improving functional mobility. In PLS, stretching is thought to improve flexibility and can also reduce muscle spasticity and cramps.[5]
Patients with PLS may find it beneficial to have an evaluation, as well as follow-up visits at multidisciplinary clinics, similar to those available for people with ALS. These multidisciplinary clinics may provide patients with the necessary treatment that they require by having an occupational therapist, physical therapist, speech language pathologist, dietician and nutritionist, all in one site.[4]
Prognosis
Patients can often live with PLS for many years and very often outlive their neurological disease and succumb to some unrelated condition. There is currently no effective cure, and the progression of symptoms varies. Some people may retain the ability to walk without assistance, but others eventually require wheelchairs, canes, or other assistive devices.
References
- RESERVED, INSERM US14-- ALL RIGHTS. "Orphanet: Primary lateral sclerosis". www.orpha.net. Retrieved 18 May 2019.
- "OMIM Entry - % 611637 - PRIMARY LATERAL SCLEROSIS, ADULT, 1; PLSA1". omim.org. Retrieved 5 August 2017.
- "Motor neuron diseases". Archived from the original on April 28, 2009. Retrieved 2009-06-02.
- Primary Lateral Sclerosis at eMedicine
- "Primary Lateral Sclerosis". Spastic Paraplegia Foundation. February 15, 2009. Archived from the original on January 16, 2013. Retrieved 2011-05-11.
- Grace, G. M.; Orange, J. B.; Rowe, A; Findlater, K; Freedman, M; Strong, M. J. (2011). "Neuropsychological functioning in PLS: A comparison with ALS". The Canadian Journal of Neurological Sciences. 38 (1): 88–97. doi:10.1017/S0317167100120803. PMID 21156436. INIST:23743303.
- Rollins, Yvonne D.; Oskarsson, Björn; Ringel, Steven P. (2011). "Primary lateral sclerosis". In Lisak, Robert; Truong, Daniel; Carroll, William; Bhidayasiri, Roongroj (eds.). International Neurology. John Wiley & Sons. pp. 203–4. ISBN 978-1-4443-1701-5.
- Tartaglia, Maria Carmela; Rowe, Ann; Findlater, Karen; Orange, J. B.; Grace, Gloria; Strong, Michael J. (2007). "Differentiation Between Primary Lateral Sclerosis and Amyotrophic Lateral Sclerosis". Archives of Neurology. 64 (2): 232–6. doi:10.1001/archneur.64.2.232. PMID 17296839.
- Gotkine, Marc; Argov, Zohar (2007). "Clinical Differentiation Between Primary Lateral Sclerosis and Upper Motor Neuron Predominant Amyotrophic Lateral Sclerosis". Archives of Neurology. 64 (10): 1545, author reply 1545. doi:10.1001/archneur.64.10.1545-a. PMID 17923644.
- "ICD-11 - Mortality and Morbidity Statistics". icd.who.int.
- http://www.mayoclinic.org/diseases-conditions/primary-lateral-sclerosis/symptoms-causes/dxc-20214456%5B%5D
- "Primary Lateral Sclerosis Information Page". National Institute of Neurological Disorders and Stroke. April 23, 2010. Archived from the original on June 6, 2011. Retrieved 2011-05-11.
- Pringle, C. E.; Hudson, A. J.; Munoz, D. G.; Kiernan, J. A.; Brown, W. F.; Ebers, G. C. (1992). "Primary Lateral Sclerosis: Clinical Features, Neuropathology and Diagnostic Criteria". Brain. 115 (2): 495–520. doi:10.1093/brain/115.2.495. PMID 1606479.
- Floeter, Mary Kay; Mills, Reversa (2009). "Progression in primary lateral sclerosis: A prospective analysis". Amyotrophic Lateral Sclerosis. 10 (5–6): 339–46. doi:10.3109/17482960903171136. PMC 3434688. PMID 19922121.
External links
| Classification | |
|---|---|
| External resources |