Glutaric aciduria type 1
Glutaric acidemia type 1 is an inherited disorder in which the body is unable to completely break down the amino acids lysine, hydroxylysine and tryptophan. Excessive levels of their intermediate breakdown products (glutaric acid, glutaryl-CoA, 3-hydroxyglutaric acid, glutaconic acid) can accumulate and cause damage to the brain (and also other organs[1]), but particularly the basal ganglia, which are regions that help regulate movement. GA1 causes secondary carnitine deficiency, as glutaric acid, like other organic acids, is detoxified by carnitine. Mental retardation may also occur.
| Glutaric acidemia type 1 | |
|---|---|
| Other names | Glutaric aciduria, GA1, GAT1 |
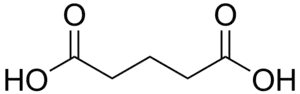 | |
| Glutaric acid | |
| Specialty | Endocrinology |
Signs and symptoms
The severity of glutaric acidemia type 1 varies widely; some individuals are only mildly affected, while others suffer severe problems. GA1 can be defined as two clinical entities: GA-1 diagnosed at birth or pre-birth and managed through dietary restrictions, and GA-1 diagnosed after an encephalopathic crisis. A crisis may occur under both headings, but individuals diagnosed prior to a crisis can be managed to avoid most or all injury.
Macrocephaly
Babies with glutaric acidemia type 1 often are born with unusually large heads (macrocephaly). Macrocephaly is amongst the earliest signs of GA1. It is thus important to investigate all cases of macrocephaly of unknown origins for GCDH deficiency,[2][3] given the importance of the early diagnosis of GA1.[4] Macrocephaly is a "pivotal clinical sign" of many neurological diseases. Physicians and parents should be aware of the benefits of investigating for an underlying neurological disorder, particularly a neurometabolic one, in children with head circumferences in the highest percentiles.
Neuromotor aspects
Affected individuals may have difficulty moving and may experience spasms, jerking, rigidity or decreased muscle tone and muscle weakness (which may be the result of secondary carnitine deficiency). Glutaric aciduria type 1, in cases of suffered crisis, can be defined as a cerebral palsy of genetic origins.
Occupational therapy

A common way to manage striatal necrosis is to provide special seating. These special wheelchairs are designed to limit abnormal movements. However, spasticity can be worsened by constraint.
Parents and caregivers can provide a more interactive occupational therapy by enabling the child to use his or her own excessive postural muscle tone to his or her own advantage (see picture; note the care with which minimal pressure is applied while ensuring safety).
The excessive tone can also be managed with "jolly jumpers" and other aids to the upright stance that do not constrain the child but help him or her gradually tone down the rigidity. Lateral sulcus becomes non operculated.
Bleeding abnormalities
Some individuals with glutaric acidemia have developed bleeding in the brain or eyes that could be mistaken for the effects of child abuse.
Genetics
The condition is inherited in an autosomal recessive pattern: mutated copies of the gene GCDH must be provided by both parents to cause glutaric acidemia type 1. The GCDH gene encodes the enzyme glutaryl-CoA dehydrogenase. This enzyme is involved in degrading the amino acids lysine, hydroxylysine and tryptophan. Mutations in the GCDH' gene prevent production of the enzyme or result in the production of a defective enzyme with very low residual activity, or an enzyme with relatively high residual activity but still phenotypic consequences.[5][6] This enzyme deficiency allows glutaric acid, 3-hydroxyglutaric acid and (to a lesser extent) glutaconic acid to build up to abnormal levels, especially at times when the body is under stress. These intermediate breakdown products are particularly prone to affect the basal ganglia, causing many of the signs and symptoms of glutaric acidemia type 1.
Glutaric acidemia type 1 occurs in approximately 1 of every 30,000 to 40,000 births. It is much more common in the Amish community and in the Ojibway population of Canada, where up to 1 in 300 newborns may be affected.
Relatives of children with GA1 can have very low GCDH activity: in an early study of GA1, GCDH activity was found to be 38%, 42%, and 42% of controls in three of the four relatives tested.[7] Those levels are close to those found by Christensen & al[5] in some heavily symptomatic GA1-affected children.
Diagnosis
Normally in MRI the Sylvian fissure is operculated, but in glutaric acidemia type 1, it is not operculated. In many areas, GA1 is included in newborn screening panels. Elevated glutarylcarnitine can be detected by mass spectrometry in a dried blood spot collected shortly after birth. After a positive screening result, confirmatory testing is performed. This includes urine organic acid analysis, looking for glutaric acid and 3-hydroxyglutaric acid. Plasma and urine acylcarnitine analysis can also be informative. Molecular analysis, including gene sequencing and copy number analysis of GCDH can be performed to confirm the diagnosis. Molecular testing can also provide information for family planning and prenatal testing, if desired.
Treatment
Correction of secondary carnitine depletion
Like many other organic acidemias, GA1 causes carnitine depletion.[8] Whole-blood carnitine can be raised by oral supplementation. However, this does not significantly change blood concentrations of glutarylcarnitine or esterified carnitine,[4] suggesting that oral supplementation is suboptimal in raising tissue levels of carnitine. In the field of clinical nutrition, researchers come to the same conclusion, that oral carnitine raises plasma levels but doesn't affect muscle carnitine, where most of it is stored and used.[9]
- In contrast, regular intravenous infusions of carnitine caused distinct clinical improvements: "decreased frequency of decompensations, improved growth, improved muscle strength and decreased reliance on medical foods with liberalization of protein intake."[8]
- Choline increases carnitine uptake and retention.[10] Choline supplements are inexpensive, safe (probably even in all children requiring anticholinergics) and can provide spectacular evidence of the suboptimal efficiency of carnitine supplementation by increasing exercise tolerance, truncal tone and general well-being.
Precursor restriction
Dietary control may help limit progression of the neurological damage.
Tryptophan
Formulas such as XLys, XTrp Analog, XLys, XTrp Maxamaid, XLys, XTrp Maxamum or Glutarex 1 are designed to provide amino acids other than lysine and tryptophan, in order to tentatively prevent protein malnutrition.
The entry of tryptophan to the brain is crucial in the proper synthesis of the neurotransmitter serotonin in the brain. One way to acutely cause depression or bulimia or anxiety in humans, in order to assess an individual's vulnerability to those disorders, is to supplement with a formula with all or most amino acids except tryptophan. The protein synthesis elicited by the amino acids leads circulating amino acids, including tryptophan, to be incorporated into proteins. Tryptophan thus lowers in the brain as a result of the protein synthesis enhancement (causing circulating tryptophan to lower more than other amino acids),[11] and perhaps also competition of large neutral amino acids for transport across the blood–brain barrier through the large neutral amino acid transporter 1 (LNAA1). The consequence is acute tryptophan depletion (ATD) in the brain and a consecutive lowering of serotonin synthesis. ATD, which is basically a diagnostic procedure, is not a treatment for GA1.
In the Amish community, where GA1 is overrepresented (Morton, 2003), patients with GA1 did not and still don't receive tryptophan-free formulas, neither as the sole source of amino acids, nor as a supplement to protein restriction. Doctor D. Holmes Morton, the 1993 Albert Schweitzer Prize for Humanitarianism laureate, is taking care of patients affected with GA1 and other metabolic diseases in this community in his Clinic for Special Children.
5-hydroxytryptophan, the precursor of serotonin that is not metabolized to glutaryl-CoA, glutaric acid and secondary metabolites, could be used as an adjunct to selective tryptophan restriction, considering the risks associated with the procedure. However, the evidence in favour of selective tryptophan restriction remains insufficient and the consensus evolves towards the restriction of lysine only.[12]
Lysine
Lysine restriction, as well as carnitine supplementation, are considered the best predictors of a good prognosis for GA1.[12] This excludes, however, patients who already suffered an encephalopathic crisis, for whom the prognosis is more related to the treatment of their acquired disorder (striatal necrosis, frontotemporal atrophy).
Protein restriction
Vegetarian diets and, for younger children, breastfeeding[13] are common ways to limit protein intake without endangering tryptophan transport to the brain.
Lysine and hydroxylysine anabolic pathway enhancement
A possible way to prevent the build-up of metabolites is to limit lysine and hydroxylysine degradation, as lysine is one of the most abundant amino acids and tryptophan is one of the least abundant amino acids.
Interaction of GCDH deficiency with GLO deficiency
While GCDH deficiency is a rare disease, GLO deficiency is the most common of metabolic diseases affecting children, limiting ascorbic acid biosynthesis to a minute fraction of what other non-primate species synthesize. It was thus called by OMIM (Online Mendeleian Inheritance in Man) a "public" error of metabolism. Ascorbic acid (Vitamin C) is a necessary cofactor for the utilization of lysine in collagen synthesis. Collagen, the most abundant protein in the human body, requires great amounts of lysine, the most abundant amino acids in proteins. Ascorbic acid, the main hydroxyl radical quencher, works as the cofactor providing the hydroxyl radical required to collagen cross-linking; lysine thus becomes hydroxylysine.
GA1 worsens during stresses and catabolic episodes, such as fasts and infections. Endogenous catabolism of proteins could be an important route for glutaric acid production. It thus follows that collagen breakdown (and protein breakdown in general) should be prevented by all possible means.
Ascorbic acid is used to prevent multiple organ failure and to lessen mortality and morbidity in intensive care units.[14] It thus appears reasonable to include sufficient doses of ascorbic acid to the treatment protocol during stresses and other challenges to growth in order to stimulate collagen synthesis and thus prevent lysine breakdown.
Tryptophan anabolic pathway enhancement
The conversion of tryptophan to serotonin and other metabolites depends on vitamin B6.[15] If tryptophan catabolism has any impact on brain glutaric acid and other catabolite levels, vitamin B6 levels should be routinely assayed and normalized in the course of the treatment of GA1.
Management of intercurrent illnesses
Stress caused by infection, fever or other demands on the body may lead to worsening of the signs and symptoms, with only partial recovery.
Prognosis
A 2006 study of 279 patients found that of those with symptoms (185, 66%), 95% had suffered an encephalopathic crises usually with following brain damage. Of the persons in the study, 49 children died and the median age of death was 6.6 years. A Kaplan-Meier analysis of the data estimated that about 50% of symptomatic cases would die by the age of 25.[12] More recent studies provide an updated prognosis where individuals affected can, through proper dietary management and carnitine supplementation, manage the disease with a much improved prognosis. Newborn screening has allowed affected patients to avoid crises and live full lives without any injury to the brain. It is essential that victims of the disease be diagnosed at birth or pre-birth and that all variables be strictly managed in order to maintain quality of life. When suspected and in the absence of confirmed diagnosis (through genetic sequencing), it is critical that the individual maintain a diet restrictive of all proteins and that blood sugars be monitored rigorously. The WHO now considers this disease entirely manageable.[16]
Epidemiology
GA1 can be described as a metabolic disorder, a neurometabolic disease, a cerebral palsy or a basal ganglia disorder (it is also misdiagnosed as shaken baby syndrome). Depending on the paradigm adopted, GA1 will mostly be managed with precursor restriction or with neurorehabilitation.
So-called "orphan diseases", such as GA1, can be adopted into wider groups of diseases (such as carnitine deficiency diseases, cerebral palsies of diverse origins, basal ganglia disorders, and others); Morton at al. (2003b) emphasize that acute striatal necrosis is a distinctive pathologic feature of at least 20 other disorders of very different etiologies (e.g. HIV encephalopathy-AIDS dementia complex, pneumococcal meningitis, hypoadrenal crisis, methylmalonic acidemia, propionic acidemia, middle cerebral artery occlusion, hypertensive vasculopathy, acute Mycoplasma pneumoniae infection, 3-nitropropionic acid intoxication, late onset familial dystonia, cerebrovascular abrupt and severe neonatal asphyxia ("selective neuronal necrosis")).
Amongst 279 patients who had been reported to have GA1, 185 were symptomatic (two thirds); being symptomatic was seen as an indication of "low treatment efficacy". High risk screening, neonatal screening and a diagnosis of macrocephaly were the ways to identify bearers of the GCDH' defective gene who weren't frankly symptomatic. Macrocephaly remains the main sign of GA1 for those who aren't related to GA1 in any way or benefit from no screening program. GA1 was considered as a "treatable disease".[12] Two thirds of the patients who have GA1 will receive little benefit from the treatment for GA1 but can benefit from treatments given to victims of middle cerebral artery occlusion, AIDS dementia and other basal ganglia disorders: brain implants, stem cell neurorestoration, growth factors, monoaminergic agents, and many other neurorehabilitation strategies.
References
- Chow, S. L.; Rohan, C.; Morris, A. A. M.; Morris, A. A. M. (2003). "Case Report: Rhabdomyolysis in Glutaric Aciduria Type I". Journal of Inherited Metabolic Disease. 26 (7): 711–712. doi:10.1023/b:boli.0000005635.89043.8a. PMID 14707521.
- Mahfoud Hawilou, Antonieta; Domínguez Méndez, Carmen Luisa; Rizzo, Cristiano; Ribes Rubio, Antonia (2004). "Macrocefalia in utero como manifestación clínica de aciduria glutárica tipo I. Informe de una nueva mutación" [In Utero Macrocephaly as Clinical Manifestation of Glutaric Aciduria Type I. Report of a Novel Mutation]. Revista de Neurología (in Spanish). 39 (10): 939. doi:10.33588/rn.3910.2004258. PMID 15573311.
- Martínez Granero, MA; Garcia Pérez, A; Martínez-Pardo, M; Parra, E (2005). "Macrocefalia como forma de presentación de la aciduria glutárica tipo 1. Importancia de un diagnóstico precoz" [Macrocephaly the first manifestation of glutaric aciduria type I: the importance of early diagnosis]. Neurología. 20 (5): 255–260. PMID 15954035.
- Strauss, Kevin A.; Puffenberger, Erik G.; Robinson, Donna L.; Morton, D. Holmes (15 August 2003). "Type I glutaric aciduria, part 1: Natural history of 77 patients". American Journal of Medical Genetics. 121C (1): 38–52. doi:10.1002/ajmg.c.20007. PMID 12888985.
- Christensen E, Aracil A, Vilaseca MA, Busquets C, Ribes A, Pineda M (1998). "Glutaric aciduria type I with high residual glutaryl-CoA dehydrogenase activity". Dev Med Child Neurol. 40 (12): 840–2. doi:10.1111/j.1469-8749.1998.tb12362.x. PMID 9881681.
- Christensen, E.; Ribes, A.; Merinero, B.; Zschocke, J. (2004). "Correlation of genotype and phenotype in glutaryl-CoA dehydrogenase deficiency". Journal of Inherited Metabolic Disease. 27 (6): 861–868. doi:10.1023/B:BOLI.0000045770.93429.3c. PMID 15505393.
- Whelan, DT; Hill, R; Ryan, ED; Spate, M (January 1979). "L-Glutaric acidemia: investigation of a patient and his family". Pediatrics. 63 (1): 88–93. PMID 440804.
- Winter, S. C. (2003). "Treatment of carnitine deficiency". Journal of Inherited Metabolic Disease. 26 (2): 171–180. doi:10.1023/a:1024433100257. PMID 12889658.
- Brass, Eric P (August 2000). "Supplemental carnitine and exercise". The American Journal of Clinical Nutrition. 72 (2): 618S–623S. doi:10.1093/ajcn/72.2.618S. PMID 10919968.
- Daily, James W.; Sachan, Dileep S. (July 1995). "Choline Supplementation Alters Carnitine Homeostasis in Humans and Guinea Pigs". The Journal of Nutrition. 125 (7): 1938–1944. doi:10.1093/jn/125.7.1938. PMID 7616311.
- Young SN (1993). "The use of diet and dietary components in the study of factors controlling affect in humans: a review". J Psychiatry Neurosci. 18 (5): 235–44. PMC 1188544. PMID 8297922.
- Kölker, Stefan; Garbade, Sven F; Greenberg, Cheryl R; Leonard, James V; Saudubray, Jean-Marie; Ribes, Antonia; Kalkanoglu, H Serap; Lund, Allan M; Merinero, Begoña; Wajner, Moacir; Troncoso, Mónica; Williams, Monique; Walter, John H; Campistol, Jaume; MartÍ-Herrero, Milagros; Caswill, Melissa; Burlina, Alberto B; Lagler, Florian; Maier, Esther M; Schwahn, Bernd; Tokatli, Aysegul; Dursun, Ali; Coskun, Turgay; Chalmers, Ronald A; Koeller, David M; Zschocke, Johannes; Christensen, Ernst; Burgard, Peter; Hoffmann, Georg F (June 2006). "Natural History, Outcome, and Treatment Efficacy in Children and Adults with Glutaryl-CoA Dehydrogenase Deficiency". Pediatric Research. 59 (6): 840–847. doi:10.1203/01.pdr.0000219387.79887.86. PMID 16641220.
- Gokcay, G.; Baykal, T.; Gokdemir, Y.; Demirkol, M. (April 2006). "Breast feeding in organic acidaemias". Journal of Inherited Metabolic Disease. 29 (2–3): 304–310. doi:10.1007/s10545-005-0255-y. PMID 16763892.
- Lovat, R.; Preiser, J. C. (2003). "Antioxidant therapy in intensive care". Current Opinion in Critical Care. 9 (4): 266–270. doi:10.1097/00075198-200308000-00003. PMID 12883280.
- Hartvig, P.; Lindner, K. J.; Bjurling, P.; Långström, B.; Tedroff, J. (June 1995). "Pyridoxine effect on synthesis rate of serotonin in the monkey brain measured with positron emission tomography". Journal of Neural Transmission. 102 (2): 91–97. doi:10.1007/BF01276505. PMID 8748674.
- Boy, Nikolas; Mühlhausen, Chris; Maier, Esther M.; Heringer, Jana; Assmann, Birgit; Burgard, Peter; Dixon, Marjorie; Fleissner, Sandra; Greenberg, Cheryl R.; Harting, Inga; Hoffmann, Georg F.; Karall, Daniela; Koeller, David M.; Krawinkel, Michael B.; Okun, Jürgen G.; Opladen, Thomas; Posset, Roland; Sahm, Katja; Zschocke, Johannes; Kölker, Stefan (16 November 2016). "Proposed recommendations for diagnosing and managing individuals with glutaric aciduria type I: second revision". Journal of Inherited Metabolic Disease. 40 (1): 75–101. doi:10.1007/s10545-016-9999-9. PMID 27853989.
Further reading
- Mahfoud Hawilou, Antonieta; Domínguez Méndez, Carmen Luisa; Rizzo, Cristiano; Ribes Rubio, Antonia (2004). "Macrocefalia in utero como manifestación clínica de aciduria glutárica tipo I. Informe de una nueva mutación" [In utero macrocephaly as clinical manifestation of glutaric aciduria type I. Report of a novel mutation]. Revista de Neurología (in Spanish). 39 (10): 939–942. doi:10.33588/rn.3910.2004258. PMID 15573311.
- Martínez Granero MA, Garcia Pérez A, Martínez-Pardo M, Parra E (2005). "[Macrocephaly the first manifestation of glutaric aciduria type I: the importance of early diagnosis.]". Neurologia (in Spanish). 20 (5): 255–60. PMID 15954035.
- Strauss KA, Morton DH (2003). "Type I glutaric aciduria, part 2: a model of acute striatal necrosis" (PDF). Am J Med Genet C Semin Med Genet. 121C (1): 53–70. doi:10.1002/ajmg.c.20008. PMID 12888986. Archived from the original (– Scholar search) on January 30, 2005. see also Part 1 referenced above