Hermansky–Pudlak syndrome
Heřmanský–Pudlák syndrome (often written Hermansky–Pudlak syndrome or abbreviated HPS) is an extremely rare autosomal recessive[1] disorder which results in oculocutaneous albinism (decreased pigmentation), bleeding problems due to a platelet abnormality (platelet storage pool defect), and storage of an abnormal fat-protein compound (lysosomal accumulation of ceroid lipofuscin). It is considered to affect around 1 in 500,000 people worldwide, with a significantly higher occurrence in Puerto Ricans, with a prevalence of 1 in 1800.[2] Many of the clinical research studies on the disease have been conducted in Puerto Rico.
| Hermansky–Pudlak syndrome | |
|---|---|
| Other names | Albinism with hemorrhagic diathesis and pigmented reticuloendothelial cells, Delta storage pool disease |
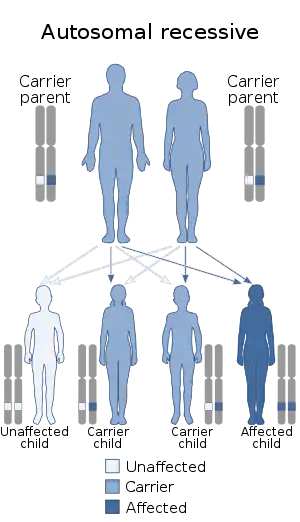 | |
| Hermansky–Pudlak syndrome is inherited via autosomal recessive manner | |
| Specialty | Endocrinology |
There are eight classic forms of the disorder, based on the genetic mutation from which the disorder stems.[3]
Signs and symptoms
There are three main disorders caused by Hermansky–Pudlak syndrome, which result in these symptoms:
- Albinism and eye problems: Individuals will have varying amounts of skin pigment (melanin). Because of the albinism there are eye problems such as light sensitivity (photophobia), strabismus (crossed eyes), and nystagmus (involuntary eye movements). Hermansky–Pudlak syndrome also impairs vision.
- Bleeding disorders: Individuals with the syndrome have platelet dysfunction. Since platelets are necessary for blood clotting, individuals will bruise and bleed easily.
- Cellular storage disorders: The syndrome causes a wax-like substance (ceroid) to accumulate in the body tissues and cause damage, especially in the lungs and kidneys.[4]
It is also associated with granulomatous colitis,, an inflammation of the colon, and with pulmonary fibrosis, a potentially fatal lung disease.
Causes
HPS can be caused by mutations in several genes: HPS1, HPS3, HPS4, HPS5, HPS6 and HPS7.
HPS type 2, which includes immunodeficiency in its phenotype, is caused by mutation in the AP3B1 gene.
HPS type 7 may result from a mutation in the gene coding for dysbindin protein.[5]
Hermansky–Pudlak syndrome is thought to be inherited as an autosomal recessive genetic trait. The defective gene, called HSP , responsible for this disorder is located on the long arm of chromosome 10 (10q2). Some research suggests that an abnormality of lysosomal function may be responsible for the development of the disease.
HPS1, AP3B1, HPS3, HPS4, HPS5, HPS6, DTNBP1 and Biogenesis of Lysosome-related Organelles Complex (BLOC1, BLOC1S2 and BLOC3) are associated with Hermansky–Pudlak syndrome.
In autosomal recessive disorders, the condition does not appear unless a person inherits two copies of the defective gene responsible for the disorder, one copy coming from each parent. If an individual receives one normal gene and one gene for the disorder, the person will be a carrier for the disease, but usually will not show symptoms. The risk of transmitting the disease to the children of a couple, both of whom are carriers for a recessive disorder, is 25 percent. Fifty percent of their children risk being carriers of the disease, but generally will not show symptoms of the disorder. Twenty-five percent of their children may receive both normal genes, one from each parent, and will be genetically normal (for that particular trait). The risk is the same for each pregnancy.[6]
Pathophysiology
The mechanism of Hermansky–Pudlak syndrome indicates that platelets in affected individuals accumulate abnormally with thrombin, epinephrine, and adenosine diphosphate, furthermore platelets in these individuals have a lower amount of dense bodies[7]
Diagnosis
The diagnosis of HPS is established by clinical findings of hypopigmentation of the skin and hair, characteristic eye findings, and demonstration of absent dense bodies on whole mount electron microscopy of platelets. Molecular genetic testing of the HPS1 gene is available on a clinical basis for individuals from northwestern Puerto Rico. Molecular testing of the HPS3 gene is available on a clinical basis for individuals of central Puerto Rican or Ashkenazi Jewish heritage. Sequence analysis is available on a clinical basis for mutations in HPS1 and HPS4. Diagnosis of individuals with other types of HPS is available on a research basis only.[8]
Treatment
While there is no cure for HPS, treatment for chronic hemorrhages associated with the disorder includes therapy with vitamin E and the antidiuretic dDAVP.[9]
Considerations for patients
A preoperative pulmonology consultation is needed. The anesthesia team should be aware that patients may have postoperative pulmonary complications as part of the syndrome.
Preoperative hematology consultation is advisable prior to elective ocular surgeries. Since patients with the syndrome have bleeding tendencies, intraoperative, perioperative, and postoperative hemorrhages should be prevented and treated. If platelet aggregation improves with desmopressin, it may be administered in the preoperative period. However, sometimes plasmapheresis is needed in the perioperative period.
Ophthalmologists should try to avoid retrobulbar blocks in patients with the syndrome. Whenever possible, patients with HPS may benefit from general endotracheal anesthesia. Phacoemulsification may help prevent intraoperative and postoperative bleeding in patients with the syndrome. Prolonged bleeding has been reported following strabismus surgery in patients with the syndrome. [10]
Prognosis
The course of HPS has been mild in rare instances of the disorder,[11] however, the general prognosis is still considered to be poor.
The disease can cause dysfunctions of the lungs, intestine, kidneys, and heart. The major complication of most forms of the disorder is pulmonary fibrosis, which typically exhibits in patients ages 40–50 years.[12] This is a fatal complication seen in many forms of HPS, and is the usual cause of death from the disorder.[13] HPS patients who develop pulmonary fibrosis typically have type 1 or type 4.
Research
HPS is one of the rare lung diseases currently being studied by The Rare Lung Diseases Consortium (RLDC). The RLDC is part of the Rare Diseases Clinical Research Network (RDCRN), an initiative of the Office of Rare Diseases Research (ORDR), of the National Center for Advancing Translational Sciences (NCATS). The RLDC is dedicated to developing new diagnostics and therapeutics for patients with rare lung diseases, through collaboration between the NIH, patient organizations and clinical investigators.
Society
Hermansky–Pudlak syndrome patients, families, and caregivers are encouraged to join the NIH Rare Lung Diseases Consortium Contact Registry. This is a privacy protected site that provides up-to-date information for individuals interested in the latest scientific news, trials, and treatments related to rare lung diseases.
See also
- Biogenesis of lysosome-related organelles complex 1
- List of cutaneous conditions
References
- Oh, J; Ho, L; Ala-Mello, S; Amato, D; Armstrong, L; Bellucci, S; Carakushansky, G; Ellis, Jp; Fong, Ct; Green, Js; Heon, E; Legius, E; Levin, Av; Nieuwenhuis, Hk; Pinckers, A; Tamura, N; Whiteford, Ml; Yamasaki, H; Spritz, Ra (March 1998). "Mutation analysis of patients with Hermansky–Pudlak syndrome: a frameshift hot spot in the HPS gene and apparent locus heterogeneity". American Journal of Human Genetics. 62 (3): 593–8. doi:10.1086/301757. PMC 1376951. PMID 9497254.
- Santiago Borrero PJ, Rodríguez-Pérez Y, Renta JY, et al. (January 2006). "Genetic testing for oculocutaneous albinism type 1 and 2 and Hermansky–Pudlak syndrome type 1 and 3 mutations in Puerto Rico". J. Invest. Dermatol. 126 (1): 85–90. doi:10.1038/sj.jid.5700034. PMC 3560388. PMID 16417222.
- Online Mendelian Inheritance in Man (OMIM): 203300
- "Hermansky–Pudlak Syndrome". Retrieved 24 November 2008.
- Li W, Zhang Q, Oiso N, Novak EK, Gautam R, O'Brien EP, Tinsley CL, Blake DJ, Spritz RA, Copeland NG, Jenkins NA, Amato D, Roe BA, Starcevic M, Dell'Angelica EC, Elliott RW, Mishra V, Kingsmore SF, Paylor RE, Swank RT (2003). "Hermansky–Pudlak syndrome type 7 (HPS-7) results from mutant dysbindin, a member of the biogenesis of lysosome-related organelles complex 1 (BLOC-1)". Nat. Genet. 35 (1): 84–9. doi:10.1038/ng1229. PMC 2860733. PMID 12923531.
- "CIGNA - Hermansky–Pudlak Syndrome". Retrieved 24 November 2008.
- "Hermansky-Pudlak Syndrome: Background, Pathophysiology, Epidemiology". 13 June 2017. Cite journal requires
|journal=(help) - Hermansky–Pudlak Syndrome. University of Washington, Seattle. 1993. Retrieved 24 November 2008.
- Wijermans, Pw; Van Dorp, Db (March 1989). "Hermansky–Pudlak syndrome: correction of bleeding time by 1-desamino-8D-arginine vasopressin". American Journal of Hematology. 30 (3): 154–7. doi:10.1002/ajh.2830300307. ISSN 0361-8609. PMID 2916560.
- "Hermansky–Pudlak Syndrome". Retrieved 24 November 2008.
- Schallreuter, Ku; Frenk, E; Wolfe, Ls; Witkop, Cj; Wood, Jm (1993). "Hermansky–Pudlak syndrome in a Swiss population" (Free full text). Dermatology. 187 (4): 248–56. doi:10.1159/000247258. ISSN 1018-8665. PMID 8274781.
- Depinho, Ra; Kaplan, Kl (May 1985). "The Hermansky–Pudlak syndrome. Report of three cases and review of pathophysiology and management considerations". Medicine. 64 (3): 192–202. doi:10.1097/00005792-198505000-00004. ISSN 0025-7974. PMID 3921802. S2CID 20833320.
- Davies, Bh; Tuddenham, Eg (April 1976). "Familial pulmonary fibrosis associated with oculocutaneous albinism and platelet function defect. A new syndrome". The Quarterly Journal of Medicine. 45 (178): 219–32. ISSN 0033-5622. PMID 940919.
- synd/2220 at Who Named It?
- Hermansky, F; Pudlak, P (1 February 1959). "Albinism associated with hemorrhagic diathesis and unusual pigmented reticular cells in the bone marrow: report of two cases with histochemical studies" (Free full text). Blood. 14 (2): 162–9. doi:10.1182/blood.V14.2.162.162. ISSN 0006-4971. PMID 13618373.
- Khalid Al Aboud; Daifullah Al Aboud (19 June 2013). "EPONYMS IN THE DERMATOLOGY LITERATURE LINKED TO CZECH REPUBLIC" (Free full text). Our Dermatology. 4 (2): 426–8.
External links
| Classification | |
|---|---|
| External resources |