Replisome
The replisome is a complex molecular machine that carries out replication of DNA. The replisome first unwinds double stranded DNA into two single strands. For each of the resulting single strands, a new complementary sequence of DNA is synthesized. The net result is formation of two new double stranded DNA sequences that are exact copies of the original double stranded DNA sequence.[1]
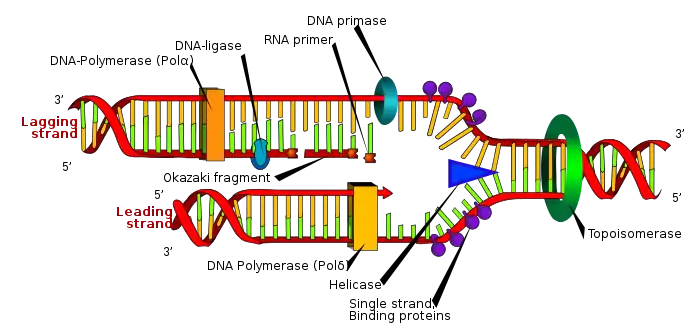
In terms of structure, the replisome is composed of two replicative polymerase complexes, one of which synthesizes the leading strand, while the other synthesizes the lagging strand. The replisome is composed of a number of proteins including helicase, RFC, PCNA, gyrase/topoisomerase, SSB/RPA, primase, DNA polymerase III, RNAse H, and ligase.
Overview of prokaryotic DNA replication process
For prokaryotes, each dividing nucleoid (region containing genetic material which is not a nucleus) requires two replisomes for bidirectional replication. The two replisomes continue replication at both forks in the middle of the cell. Finally, as the termination site replicates, the two replisomes separate from the DNA. The replisome remains at a fixed, midcell location in the cell, attached to the membrane, and the template DNA threads through it. DNA is fed through the stationary pair of replisomes located at the cell membrane.
Overview of eukaryotic DNA replication process
For eukaryotes, numerous replication bubbles form at origins of replication throughout the chromosome. As with prokaryotes, two replisomes are required, one at each replication fork located at the terminus of the replication bubble. Because of significant differences in chromosome size, and the associated complexities of highly condensed chromosomes, various aspects of the DNA replication process in eukaryotes, including the terminal phases, are less well-characterised than for prokaryotes.
Challenges of DNA replication
The replisome is a system in which various factors work together to solve the structural and chemical challenges of DNA replication. Chromosome size and structure varies between organisms, but since DNA molecules are the reservoir of genetic information for all forms of life, many replication challenges and solutions are the same for different organisms. As a result, the replication factors that solve these problems are highly conserved in terms of structure, chemistry, functionality, or sequence. General structural and chemical challenges include the following:
- Efficient replisome assembly at origins of replication (origin recognition complexes or specific replication origin sequences in some organisms)
- Separating the duplex into the leading and lagging template strands (helicases)
- Protecting the leading and lagging strands from damage after duplex separation (SSB and RPA factors)
- Priming of the leading and lagging template strands (primase or DNA polymerase alpha)
- Ensuring processivity (clamp loading factors, ring-shaped clamp proteins, strand binding proteins)
- High-fidelity DNA replication (DNA polymerase III, DNA polymerase delta, DNA polymerase epsilon. All have intrinsically low error rates because of their structure and chemistry.)
- Error correction (replicative polymerase active sites sense errors; 3' to 5' exonuclease domains of replicative polymerases fix errors)
- Synchronised polymerisation of leading and lagging strands despite anti-parallel structure (replication fork structure, dimerisation of replicative polymerases)
- Primer removal (DNA polymerase I, RNAse H, flap endonucleases such as FEN1, or other DNA repair factors)
- Formation of phosphodiester bonds at gaps between Okazaki fragments (ligase)
In general, the challenges of DNA replication involve the structure of the molecules, the chemistry of the molecules, and, from a systems perspective, the underlying relationships between the structure and the chemistry.
Solving the challenges of DNA replication
Many of the structural and chemical problems associated with DNA replication are managed by molecular machinery that is highly conserved across organisms. This section discusses how replisome factors solve the structural and chemical challenges of DNA replication.
Replisome assembly
DNA replication begins at sites called origins of replication. In organisms with small genomes and simple chromosome structure, such as bacteria, there may be only a few origins of replication on each chromosome. Organisms with large genomes and complex chromosome structure, such as humans, may have hundreds, or even thousands, of origins of replication spread across multiple chromosomes.
DNA structure varies with time, space, and sequence, and it is thought that these variations, in addition to their role in gene expression, also play active roles in replisome assembly during DNA synthesis. Replisome assembly at an origin of replication is roughly divided into three phases.
For prokaryotes:
- Formation of pre-replication complex. DnaA binds to the origin recognition complex and separates the duplex. This attracts DnaB helicase and DnaC, which maintain the replication bubble.
- Formation of pre-initiation complex. SSB binds to the single strand and then gamma (clamp loading factor) binds to SSB.
- Formation of initiation complex. Gamma deposits the sliding clamp (beta) and attracts DNA polymerase III.
For eukaryotes:
- Formation of pre-replication complex. MCM factors bind to the origin recognition complex and separate the duplex, forming a replication bubble.
- Formation of pre-initiation complex. Replication protein A (RPA) binds to the single stranded DNA and then RFC (clamp loading factor) binds to RPA.
- Formation of initiation complex. RFC deposits the sliding clamp (PCNA) and attracts DNA polymerases such as alpha (α), delta (δ), epsilon (ε).
For both prokaryotes and eukaryotes, the next stage is generally referred to as 'elongation', and it is during this phase that the majority of DNA synthesis occurs.
Separating the duplex
DNA is a duplex formed by two anti-parallel strands. Following Meselson-Stahl, the process of DNA replication is semi-conservative, whereby during replication the original DNA duplex is separated into two daughter strands (referred to as the leading and lagging strand templates). Each daughter strand becomes part of a new DNA duplex. Factors generically referred to as helicases unwind the duplex.
Helicases
Helicase is an enzyme which breaks hydrogen bonds between the base pairs in the middle of the DNA duplex. Its doughnut like structure wraps around DNA and separates the strands ahead of DNA synthesis. In eukaryotes, the Mcm2-7 complex acts as a helicase, though which subunits are required for helicase activity is not entirely clear.[2] This helicase translocates in the same direction as the DNA polymerase (3' to 5' with respect to the template strand). In prokaryotic organisms, the helicases are better identified and include dnaB, which moves 5' to 3' on the strand opposite the DNA polymerase.
Unwinding supercoils and decatenation

As helicase unwinds the double helix, topological changes induced by the rotational motion of the helicase lead to supercoil formation ahead of the helicase (similar to what happens when you twist a piece of thread).
Gyrase and topoisomerases
Gyrase (a form of topoisomerase) relaxes and undoes the supercoiling caused by helicase. It does this by cutting the DNA strands, allowing it to rotate and release the supercoil, and then rejoining the strands. Gyrase is most commonly found upstream of the replication fork, where the supercoils form.
Protecting the leading and lagging strands
Single-stranded DNA is highly unstable and can form hydrogen bonds with itself that are referred to as 'hairpins' (or the single strand can improperly bond to the other single strand). To counteract this instability, single-strand binding proteins (SSB in prokaryotes and Replication protein A in eukaryotes) bind to the exposed bases to prevent improper ligation.
If you consider each strand as a "dynamic, stretchy string", the structural potential for improper ligation should be obvious.
| The lagging strand without binding proteins. |
|---|
|
An expanded schematic reveals the underlying chemistry of the problem: the potential for hydrogen bond formation between unrelated base pairs.
| Schematic view of newly separated DNA strands without strand binding proteins. |
|---|
|
Binding proteins stabilise the single strand and protected the strand from damage caused by unlicensed chemical reactions.
| The lagging strand coated with binding proteins (*) that prevent improper ligation. |
|---|
|
The combination of a single strand and its binding proteins serves as a better substrate for replicative polymerases than a naked single strand (binding proteins provide extra thermodynamic driving force for the polymerisation reaction). Strand binding proteins are removed by replicative polymerases.
Priming the leading and lagging strands
From both a structural and chemical perspective, a single strand of DNA by itself (and the associated single strand binding proteins) is not suitable for polymerisation. This is because the chemical reactions catalysed by replicative polymerases require a free 3' OH in order to initiate nucleotide chain elongation. In terms of structure, the conformation of replicative polymerase active sites (which is highly related to the inherent accuracy of replicative polymerases) means these factors cannot start chain elongation without a pre-existing chain of nucleotides, because no known replicative polymerase can start chain elongation de novo.
Priming enzymes, (which are DNA-dependent RNA polymerases), solve this problem by creating an RNA primer on the leading and lagging strands. The leading strand is primed once, and the lagging strand is primed approximately every 1000 (+/- 200) base pairs (one primer for each Okazaki fragment on the lagging strand). Each RNA primer is approximately 10 bases long.
| Single strand of DNA with strand binding proteins (*) and RNA primer added by priming enzymes (UAGCUAUAUAUA). |
|---|
|
The interface at (A*) contains a free 3' OH that is chemically suitable for the reaction catalysed by replicative polymerases, and the "overhang" configuration is structurally suitable for chain elongation by a replicative polymerase. Thus, replicative polymerases can begin chain elongation at (A*).
Primase
In prokaryotes, the primase creates an RNA primer at the beginning of the newly separated leading and lagging strands.
DNA polymerase alpha
In eukaryotes, DNA polymerase alpha creates an RNA primer at the beginning of the newly separated leading and lagging strands, and, unlike primase, DNA polymerase alpha also synthesizes a short chain of deoxynucleotides after creating the primer.
Ensuring processivity and synchronisation
Processivity refers to both speed and continuity of DNA replication, and high processivity is a requirement for timely replication. High processivity is in part ensured by ring-shaped proteins referred to as 'clamps' that help replicative polymerases stay associated with the leading and lagging strands. There are other variables as well: from a chemical perspective, strand binding proteins stimulate polymerisation and provide extra thermodynamic energy for the reaction. From a systems perspective, the structure and chemistry of many replisome factors (such as the AAA+ ATPase features of the individual clamp loading sub-units, along with the helical conformation they adopt), and the associations between clamp loading factors and other accessory factors, also increases processivity.
To this point, according to research by Kuriyan et al.,[3] due to their role in recruiting and binding other factors such as priming enzymes and replicative polymerases, clamp loaders and sliding clamps are at the heart of the replisome machinery. Research has found that clamp loading and sliding clamp factors are absolutely essential to replication, which explains the high degree of structural conservation observed for clamp loading and sliding clamp factors. This architectural and structural conservation is seen in organisms as diverse as bacteria, phages, yeast, and humans. That such a significant degree of structural conservation is observed without sequence homology further underpins the significance of these structural solutions to replication challenges.
Clamp loader
Clamp loader is a generic term that refers to replication factors called gamma (prokaryotes) or RFC (eukaryotes). The combination of template DNA and primer RNA is referred to as 'A-form DNA' and it is thought that clamp loading replication proteins (helical heteropentamers) want to associate with A-form DNA because of its shape (the structure of the major/minor groove) and chemistry (patterns of hydrogen bond donors and acceptors).[3][4] Thus, clamp loading proteins associate with the primed region of the strand which causes hydrolysis of ATP and provides energy to open the clamp and attach it to the strand.[3][4]
Sliding clamp
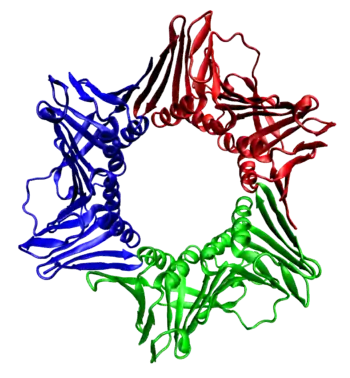
Sliding clamp is a generic term that refers to ring-shaped replication factors called beta (prokaryotes) or PCNA (eukaryotes). Clamp proteins attract and tether replicative polymerases, such as DNA polymerase III, in order to extend the amount of time that a replicative polymerase stays associated with the strand. From a chemical perspective, the clamp has a slightly positive charge at its centre that is a near perfect match for the slightly negative charge of the DNA strand.
In some organisms, the clamp is a dimer, and in other organisms the clamp is a trimer. Regardless, the conserved ring architecture allows the clamp to enclose the strand.
Dimerisation of replicative polymerases
Replicative polymerases form an asymmetric dimer at the replication fork by binding to sub-units of the clamp loading factor. This asymmetric conformation is capable of simultaneously replicating the leading and lagging strands, and the collection of factors that includes the replicative polymerases is generally referred to as a holoenzyme. However, significant challenges remain: the leading and lagging strands are anti-parallel. This means that nucleotide synthesis on the leading strand naturally occurs in the 5' to 3' direction. However, the lagging strand runs in the opposite direction and this presents quite a challenge since no known replicative polymerases can synthesise DNA in the 3' to 5' direction.
The dimerisation of the replicative polymerases solves the problems related to efficient synchronisation of leading and lagging strand synthesis at the replication fork, but the tight spatial-structural coupling of the replicative polymerases, while solving the difficult issue of synchronisation, creates another challenge: dimerisation of the replicative polymerases at the replication fork means that nucleotide synthesis for both strands must take place at the same spatial location, despite the fact that the lagging strand must be synthesised backwards relative to the leading strand. Lagging strand synthesis takes place after the helicase has unwound a sufficient quantity of the lagging strand, and this "sufficient quantity of the lagging strand" is polymerised in discrete nucleotide chains called Okazaki fragments.
Consider the following: the helicase continuously unwinds the parental duplex, but the lagging strand must be polymerised in the opposite direction. This means that, while polymerisation of the leading strand proceeds, polymerisation of the lagging strand only occurs after enough of the lagging strand has been unwound by the helicase. At this point, the lagging strand replicative polymerase associates with the clamp and primer in order to start polymerisation. During lagging strand synthesis, the replicative polymerase sends the lagging strand back toward the replication fork. The replicative polymerase disassociates when it reaches an RNA primer. Helicase continues to unwind the parental duplex, the priming enzyme affixes another primer, and the replicative polymerase reassociates with the clamp and primer when a sufficient quantity of the lagging strand has unwound.
Collectively, leading and lagging strand synthesis is referred to as being 'semidiscontinuous'.
High-fidelity DNA replication
Prokaryotic and eukaryotic organisms use a variety of replicative polymerases, some of which are well-characterised:
- DNA polymerase III
- DNA polymerase delta
- DNA polymerase epsilon
DNA polymerase III
This polymerase synthesizes leading and lagging strand DNA in prokaryotes.
DNA polymerase delta
This polymerase synthesizes lagging strand DNA in eukaryotes.[5] (Thought to form an asymmetric dimer with DNA polymerase epsilon.)[6]
DNA polymerase epsilon
This polymerase synthesizes leading strand DNA in eukaryotes.[7] (Thought to form an asymmetric dimer with DNA polymerase delta.)[5]
Proof-reading and error correction
Although rare, incorrect base pairing polymerisation does occur during chain elongation. (The structure and chemistry of replicative polymerases mean that errors are unlikely, but they do occur.) Many replicative polymerases contain a "error correction" mechanism in the form of a 3' to 5' exonuclease domain that is capable of removing base pairs from the exposed 3' end of the growing chain. Error correction is possible because base pair errors distort the position of the magnesium ions in the polymerisation sub-unit, and the structural-chemical distortion of the polymerisation unit effectively stalls the polymerisation process by slowing the reaction.[8] Subsequently, the chemical reaction in the exonuclease unit takes over and removes nucleotides from the exposed 3' end of the growing chain.[9] Once an error is removed, the structure and chemistry of the polymerisation unit returns to normal and DNA replication continues. Working collectively in this fashion, the polymerisation active site can be thought of as the "proof-reader", since it senses mismatches, and the exonuclease is the "editor", since it corrects the errors.
Base pair errors distort the polymerase active site for between 4-6 nucleotides, which means, depending on the type of mismatch, there are up to six chances for error correction.[8] The error sensing and error correction features, combined with the inherent accuracy that arises from the structure and chemistry of replicative polymerases, contributes to an error rate of approximately 1 base pair mismatch in 108 to 1010 base pairs.
| Schematic view of correct base pairs followed by 8 possible base pair mismatches.[10] |
|---|
|
Errors can be classified in three categories: purine-purine mismatches, pyrimidine-pyrimidine mismatches, and pyrimidine-purine mismatches. The chemistry of each mismatch varies, and so does the behaviour of the replicative polymerase with respect to its mismatch sensing activity.
The replication of bacteriophage T4 DNA upon infection of E. coli is a well-studied DNA replication system. During the period of exponential DNA increase at 37°C, the rate of elongation is 749 nucleotides per second.[11] The mutation rate during replication is 1.7 mutations per 108 base pairs.[12] Thus DNA replication in this system is both very rapid and highly accurate.
Primer removal and nick ligation
There are two problems after leading and lagging strand synthesis: RNA remains in the duplex and there are nicks between each Okazaki fragment in the lagging duplex. These problems are solved by a variety of DNA repair enzymes that vary by organism, including: DNA polymerase I, DNA polymerase beta, RNAse H, ligase, and DNA2. This process is well-characterised in prokaryotes and much less well-characterised in many eukaryotes.
In general, DNA repair enzymes complete the Okazaki fragments through a variety of means, including: base pair excision and 5' to 3' exonuclease activity that removes the chemically unstable ribonucleotides from the lagging duplex and replaces them with stable deoxynucleotides. This process is referred to as 'maturation of Okazaki fragments', and ligase (see below) completes the final step in the maturation process.
| RNA-DNA duplex with ribonucleotides added by a priming enzyme (-) and deoxynucleotides added by a replicative polymerase (+). |
|---|
|
Primer removal and nick ligation can be thought of as DNA repair processes that produce a chemically-stable, error-free duplex. To this point, with respect to the chemistry of an RNA-DNA duplex, in addition to the presence of uracil in the duplex, the presence of ribose (which has a reactive 2' OH) tends to make the duplex much less chemically-stable than a duplex containing only deoxyribose (which has a non-reactive 2' H).
DNA polymerase I
DNA polymerase I is an enzyme that repairs DNA.
RNAse H
RNAse H is an enzyme that removes RNA from an RNA-DNA duplex.
Ligase
After DNA repair factors replace the ribonucleotides of the primer with deoxynucleotides, a single gap remains in the sugar-phosphate backbone between each Okazaki fragment in the lagging duplex. An enzyme called DNA ligase connects the gap in the backbone by forming a phosphodiester bond between each gap that separates the Okazaki fragments. The structural and chemical aspects of this process, generally referred to as 'nick translation', exceed the scope of this article.
| A schematic view of the new, lagging strand daughter DNA duplex is shown below, along with the sugar-phosphate backbone. |
|---|
|
| The finished duplex: |
|---|
|
Replication stress
Replication stress can result in a stalled replication fork. One type of replicative stress results from DNA damage such as inter-strand cross-links (ICLs). An ICL can block replicative fork progression due to failure of DNA strand separation. In vertebrate cells, replication of an ICL-containing chromatin template triggers recruitment of more than 90 DNA repair and genome maintenance factors.[13] These factors include proteins that perform sequential incisions and homologous recombination.
History
Katherine Lemon and Alan Grossman showed using Bacillus subtilis that replisomes do not move like trains along a track but DNA is actually fed through a stationary pair of replisomes located at the cell membrane. In their experiment, the replisomes in B. subtilis were each tagged with green fluorescent protein, and the location of the complex was monitored in replicating cells using fluorescence microscopy. If the replisomes moved like a train on a track, the polymerase-GFP protein would be found at different positions in each cell. Instead, however, in every replicating cell, replisomes were observed as distinct fluorescent foci located at or near midcell. Cellular DNA stained with a blue fluorescent dye (DAPI) clearly occupied most of the cytoplasmic space.[14]
References
- Yao, Nina Y.; O'Donnell, Mike (2010). "SnapShot: The Replisome". Cell. Elsevier BV. 141 (6): 1088–1088.e1. doi:10.1016/j.cell.2010.05.042. ISSN 0092-8674. PMC 4007198. PMID 20550941.
- Bochman ML, Schwacha A (July 2008). "The Mcm2-7 complex has in vitro helicase activity". Mol. Cell. 31 (2): 287–93. doi:10.1016/j.molcel.2008.05.020. PMID 18657510.
- Kelch BA, Makino DL, O'Donnell M, Kuriyan J (2012). "Clamp loader ATPases and the evolution of DNA replication machinery". BMC Biol. 10: 34. doi:10.1186/1741-7007-10-34. PMC 3331839. PMID 22520345.
- Bowman GD, O'Donnell M, Kuriyan J (June 2004). "Structural analysis of a eukaryotic sliding DNA clamp-clamp loader complex". Nature. 429 (6993): 724–30. doi:10.1038/nature02585. PMID 15201901.
- Swan MK, Johnson RE, Prakash L, Prakash S, Aggarwal AK (September 2009). "Structural basis of high-fidelity DNA synthesis by yeast DNA polymerase delta". Nat. Struct. Mol. Biol. 16 (9): 979–86. doi:10.1038/nsmb.1663. PMC 3055789. PMID 19718023.
- Miyabe I, Kunkel TA, Carr AM (December 2011). "The major roles of DNA polymerases epsilon and delta at the eukaryotic replication fork are evolutionarily conserved". PLOS Genet. 7 (12): e1002407. doi:10.1371/journal.pgen.1002407. PMC 3228825. PMID 22144917.
- Pursell ZF, Isoz I, Lundström EB, Johansson E, Kunkel TA (July 2007). "Yeast DNA polymerase epsilon participates in leading-strand DNA replication". Science. 317 (5834): 127–30. doi:10.1126/science.1144067. PMC 2233713. PMID 17615360.
- Johnson SJ, Beese LS (March 2004). "Structures of mismatch replication errors observed in a DNA polymerase". Cell. 116 (6): 803–16. doi:10.1016/S0092-8674(04)00252-1. PMID 15035983.
- Jiricny J (March 2004). "Unfaithful DNA polymerase caught in the act" (PDF). Mol. Cell. 13 (6): 768–9. doi:10.1016/S1097-2765(04)00149-2. PMID 15053870.
- "Mutagenesis and DNA repair". ATDBio Ltd.
- McCarthy D, Minner C, Bernstein H, Bernstein C (1976). "DNA elongation rates and growing point distributions of wild-type phage T4 and a DNA-delay amber mutant". J. Mol. Biol. 106 (4): 963–81. doi:10.1016/0022-2836(76)90346-6. PMID 789903.
- Drake JW (1970) The Molecular Basis of Mutation. Holden-Day, San Francisco ISBN 0816224501 ISBN 978-0816224500
- Räschle M, Smeenk G, Hansen RK, Temu T, Oka Y, Hein MY, Nagaraj N, Long DT, Walter JC, Hofmann K, Storchova Z, Cox J, Bekker-Jensen S, Mailand N, Mann M (2015). "DNA repair. Proteomics reveals dynamic assembly of repair complexes during bypass of DNA cross-links". Science. 348 (6234): 1253671. doi:10.1126/science.1253671. PMC 5331883. PMID 25931565.
- Foster JB, Slonczewski J (2010). Microbiology: An Evolving Science (Second ed.). New York: W. W. Norton & Company. ISBN 978-0-393-93447-2.
Further reading
- Pomerantz RT, O'Donnell M (April 2007). "Replisome mechanics: insights into a twin DNA polymerase machine". Trends Microbiol. 15 (4): 156–64. doi:10.1016/j.tim.2007.02.007. PMID 17350265.
External links
- DNA+replisome at the US National Library of Medicine Medical Subject Headings (MeSH)