Plasma cell leukemia
Plasma cell leukemia (PCL) is a plasma cell dyscrasia, i.e. a disease involving the malignant degeneration of a subtype of white blood cells called plasma cells. It is the terminal stage and most aggressive form of these dyscrasias, constituting 2% to 4% of all cases of plasma cell malignancies. PCL may present as primary plasma cell leukemia, i.e. in patients without prior history of a plasma cell dyscrasia or as secondary plasma cell dyscrasia, i.e. in patients previously diagnosed with a history of its predecessor dyscrasia, multiple myeloma. The two forms of PCL appear to be at least partially distinct from each other. In all cases, however, PCL is an extremely serious, life-threatening, and therapeutically challenging disease.[1][2]
| Plasma cell leukemia | |
|---|---|
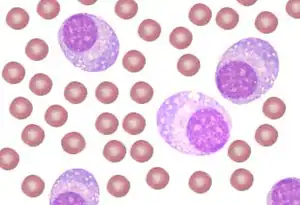 | |
| A schematic showing peripheral blood with plasma cell leukemia. Many plasma cells are seen mixed with red cells. | |
| Specialty | Hematology and oncology |
Signs and symptoms
Primary PCL
The clinical presentation of primary PCL (pPCL) indicates a far more aggressive disease than that of a typical multiple myeloma case with its clinical features being a combination of those found in multiple myeloma and acute leukemia. Like multiple myeloma patients, pPCL patients exhibit pathologically high levels of monoclonal plasma cells in their bone marrow plus a malignant plasma cell-secreted circulating monoclonal myeloma protein, either IgG, IgA, a light chain, or none in 28-56%, 4-7%, 23-44%, or 0-12% of cases, respectively.[1][3] Similar to B cell leukemias, but unlike multiple myeloma, pPCL patients exhibit relative high frequencies of splenomegaly, lymphadenopathy, hepatomegaly, kidney failure, bone marrow failure (i.e. thrombocytopenia, anemia, and/or, rarely, leukopenia), central nervous system defects, and peripheral neuropathies due to the invasion of these tissues by plasma cells and/or the deposition of their circulating monoclonal immunoglobulin in them. Compared to multiple myeloma patients, pPCL patients also: exhibit 1) high rates of developing a hypercalcemic crisis, i.e. a potentially life-threatening episode of high ionic calcium (Ca2+) levels in the blood due to excess bone re-absorption and/or renal failure; b) higher levels of serum lactate dehydrogenase and Beta-2 microglobulin; and c) lower rates of bone but higher rates of soft tissue plasma cell tumors termed plasmacytomas.[1][4]
Secondary PCL
Secondary PCL (sPCL) is diagnosed in 1-4% of patients known to have had multiple myeloma for a median time of ~21 months. It is the terminal phase of these patients myeloma disease. sPCL patients typically are highly symptomatic due to extensive disease with malignant plasma cell infiltrations in, and failures of, not only the bone marrow but also other organs. They have failed or broken through one or more treatment regimens and therefore may also show some of the toxic effects of these treatments.[1][5]
Cause
PCL is caused by the development of an excessively high number of genetic abnormalities in plasma cells or, more particularly, their precursor B cells and plasmablasts (see plasma cells). This genetic instability is due to a myriad of acquired abnormalities including gene mutations; single nucleotide polymorphisms; depletions and duplications of parts of a gene, larger portion of a chromosome, or even an entire arm of a chromosome; translocations, deletions, and duplications of entire chromosomes; and increases and decreases in the expression of intact genes due to, e.g. the methylation of gene promotors and various less direct effects. These genetic abnormalities effect the Wnt signaling pathway, regulation of the cell cycle, RNA metabolism, protein folding, and cadherin-related cell adherence to extracellular matrix. These effects in turn control plasma cell proliferation, survival, apoptosis, adhesion to bone marrow, genome stability, and secretion of monoclonal immunoglobulins.[6]
Secondary plasma cell leukemia (sPCL) results from the comparatively slow development of plasma cell/plasma cell precursor genetic abnormalities which initially create a clone of cells that cause the premalignant condition of monoclonal gammopathy of undetermined significance. In a very small percentage of these cases, the progressive development of further genetic abnormalities serially create a clone(s) of plasma cells that cause the more serious but still premalignant disorder of smoldering multiple myeloma, overt myeloma cancer, and ultimately sPCL.[7][6] In contrast to sPCL, pPCL presents de novo with a broad range of genetic abnormalities. For example, advanced methods for examining the genome viz., whole-exome sequencing and gene expression profiling, have identified 166 non-silent gene variants per pPCL patient sample at the time of diagnosis. These abnormalities are similar but not identical to those detected in sPCL while the abnormalies detected in sPCL more closely resemble those detected in multiple myeloma than do those of pPCL: the genetic data support the clinical data in suggesting that sPCL and pPCL are distinct diseases with sPCL among the two PCLs being more closely related to multiple myeloma.[6][8] Examination of plasma cell immunophenotype by measuring certain of their cell surface antigens, particularly Cluster of differentiation. CD markers on plasma cells from patients with pPCL differ from those taken form multiple myeloma or sPCL patients. For example: pPCL plasma cells more often express CD20 antigen, which is considered important in anchoring plasma cells to the bone marrow stroma, than do those on plasma cells taken from myeloma patients (50% vs. 17%); pPCL plasma cells often lack CD56 antigen which is present on the majority of plasma cells taken form multiple myeloma patients; and pPCL plasma cells more frequently express CD28 than do sPCL plasma cells. Thus, immunophenotyping supports that notion that multiple myeloma, sPCL, and pPCL show critically important fundamental differences that may explain their different clinical presentations, courses, responses to therapy, and prognoses.[6][8][9][10]
Diagnosis
The International Myeloma Working Group has defined the diagnostic criteria for plasma cell leukemia as the presence in blood of >2x109 plasma cells per liter or, alternatively, >20% of nucleated blood cells being plasma cells. More recently, the Group has suggested that values of 0.5x109 or 5%, respectively, may be more appropriate from a therapeutic viewpoint and therefore should be studied as a definitive criterion for the disease.[1] A recent study supported this suggestion in finding that multiple myeloma patients with >5% circulating plasma cells had a prognosis much worse than that for multiple myeloma and similar to that for plasma cell leukemia.[2] Flow cytometry immunophenotyping of blood cells to detect clonal phenotypes of plasma cells seen in multiple myeloma (e.g. the CD138+, CD38+, CD19−, CD45+/- phenotype) may be a more sensitive method to enumerate circulating clonal plasma cells and diagnose plasma cell leukemia.[3]
Treatments
Prior to the use of newly developed drugs and treatment regimens, median survival rates from the time of diagnosis for pPCL and sPCL were 8–11 months and 2–8 months, respectively, even when treated very aggressively with the VAD regimen of vincristine, doxorubicin, and dexamethasone or the VCMP regimen of vincristine, carmustine, melphalan, and prednisone alternating with vincristine, carmustine, doxorubicin, and prednisone.[1][5] The treatment of PCL patients, particularly pPCL patients, with newer methods appears to have made modest improvements in survival rates. However, the rarity of these two leukemias has limited individual studies to case reports on a small number of patients or rectrospective analyses of patient records. Randomized controlled trials on these patients have not been reported. One flaw of these methods is patient selection bias, i.e. patients selected for treatment with a new regimen may be less ill than average patients with the disease and therefore have an intrinsically less aggressive (i.e. longer overall survival time) disease.[4]
Primary plasma cell leukemia
Recent case report studies suggest that treatment regimens which include a proteasome inhibitor drug, particularly bortezomib, and/or autologous stem-cell transplantation have improved pPCL survival. For example, 28 patients treated with a bortezomib-based induction regimen followed by autologous stem-cell transplantation and then a maintenance regimen of lenaldomide (an immunosuppressant related to thalidomide), bortezomib, and dexamethasone (a corticosteroid) has a progression free survival rate of 66% at 3 years and an overall survival rate of 73% at 4 years. In one study, patients receiving intensive chemotherapy plus autologous stem-cell transplantation had a median survival of 34 months while those receiving chemotherapy alone had a median survival of 11 months. Two other studies that included bortezomib in their chemotherapy regimens likewise found that the addition of autologous stem-cell transplantation improved results. Current recommendations for treating pPCL often include induction with a three drug regimen such as borezomib-lenalidomide-dexamethasone followed by autologous stem-cell transplantion and consolidation/maintenance with of combination of immunomodulator agents (e.g. thalidomide, lenalidomide, or pomalidomide) plus a proteasome inhibitor (bortezomib, ixazomib, or carfilzomib.[4][10][11]
Secondary plasma cell leukemia
As the end stage of multiple myeloma that has failed or broken through one or more therapeutic regimens, sPCL continues to be highly refractory to various treatment regimens (<50%), very short response times of these regiments, and poor overall survival rates (median survival of 2-8 to months).[1][5][12] Patients with sPCL may have short-lived responses to treatment regimens (as communicated in case reports) that include bortezomid but there are no established therapeutic regimens that have clearly been shown to improve their overall or median survival.[4][10]
See also
References
- Fernández de Larrea C, Kyle RA, Durie BG, Ludwig H, Usmani S, Vesole DH, Hajek R, San Miguel JF, Sezer O, Sonneveld P, Kumar SK, Mahindra A, Comenzo R, Palumbo A, Mazumber A, Anderson KC, Richardson PG, Badros AZ, Caers J, Cavo M, LeLeu X, Dimopoulos MA, Chim CS, Schots R, Noeul A, Fantl D, Mellqvist UH, Landgren O, Chanan-Khan A, Moreau P, Fonseca R, Merlini G, Lahuerta JJ, Bladé J, Orlowski RZ, Shah JJ (2013). "Plasma cell leukemia: consensus statement on diagnostic requirements, response criteria and treatment recommendations by the International Myeloma Working Group". Leukemia. 27 (4): 780–91. doi:10.1038/leu.2012.336. PMC 4112539. PMID 23288300.
- Granell M, Calvo X, Garcia-Guiñón A, Escoda L, Abella E, Martínez CM, Teixidó M, Gimenez MT, Senín A, Sanz P, Campoy D, Vicent A, Arenillas L, Rosiñol L, Sierra J, Bladé J, de Larrea CF (2017). "Prognostic impact of circulating plasma cells in patients with multiple myeloma: implications for plasma cell leukemia definition". Haematologica. 102 (6): 1099–1104. doi:10.3324/haematol.2016.158303. PMC 5451342. PMID 28255016.
- Gonsalves, Wilson I. (2017-04-10). "Primary Plasma Cell Leukemia: A Practical Approach to Diagnosis and Clinical Management". American Journal of Hematology / Oncology. 13 (3).
- Joseph NS, Gentili S, Kaufman JL, Lonial S, Nooka AK (2017). "High-risk Multiple Myeloma: Definition and Management". Clinical Lymphoma, Myeloma & Leukemia. 17S: S80–S87. doi:10.1016/j.clml.2017.02.018. PMID 28760306.
- Rotaru I, Găman G, Dumitrescu D, Foarfă C (2012). "Secondary plasma cell leukemia". Romanian Journal of Morphology and Embryology = Revue Roumaine de Morphologie et Embryologie. 53 (4): 1073–6. PMID 23303035.
- Cifola I, Lionetti M, Pinatel E, Todoerti K, Mangano E, Pietrelli A, Fabris S, Mosca L, Simeon V, Petrucci MT, Morabito F, Offidani M, Di Raimondo F, Falcone A, Caravita T, Battaglia C, De Bellis G, Palumbo A, Musto P, Neri A (2015). "Whole-exome sequencing of primary plasma cell leukemia discloses heterogeneous mutational patterns". Oncotarget. 6 (19): 17543–58. doi:10.18632/oncotarget.4028. PMC 4627327. PMID 26046463.
- Dutta AK, Hewett DR, Fink JL, Grady JP, Zannettino AC (2017). "Cutting edge genomics reveal new insights into tumour development, disease progression and therapeutic impacts in multiple myeloma". British Journal of Haematology. 178 (2): 196–208. doi:10.1111/bjh.14649. PMID 28466550.
- Barbieri M, Manzoni M, Fabris S, Ciceri G, Todoerti K, Simeon V, Musto P, Cortelezzi A, Baldini L, Neri A, Lionetti M (2016). "Compendium of FAM46C gene mutations in plasma cell dyscrasias". British Journal of Haematology. 174 (4): 642–5. doi:10.1111/bjh.13793. PMID 26456599.
- Robillard N, Jego G, Pellat-Deceunynck C, Pineau D, Puthier D, Mellerin MP, Barillé S, Rapp MJ, Harousseau JL, Amiot M, Bataille R (1998). "CD28, a marker associated with tumoral expansion in multiple myeloma". Clinical Cancer Research. 4 (6): 1521–6. PMID 9626472.
- Jelinek T, Kryukov F, Rihova L, Hajek R (2015). "Plasma cell leukemia: from biology to treatment". European Journal of Haematology. 95 (1): 16–26. doi:10.1111/ejh.12533. PMID 25778450.
- Gonsalves WI, Rajkumar SV, Go RS, Dispenzieri A, Gupta V, Singh PP, Buadi FK, Lacy MQ, Kapoor P, Dingli D, Lust JA, Zeldenrust SR, Hayman SR, Kyle RA, Gertz MA, Kumar SK (2014). "Trends in survival of patients with primary plasma cell leukemia: a population-based analysis". Blood. 124 (6): 907–12. doi:10.1182/blood-2014-03-565051. PMC 4126330. PMID 24957143.
- Bommannan K, Sachdeva MU, Malhotra P, Kumar N, Sharma P, Naseem S, Ahluwalia J, Das R, Varma N, Prakash G, Khadwal A, Srinivasan R, Varma S (2016). "Plasma cell leukemia in North India: retrospective analysis of a distinct clinicohematological entity from a tertiary care center and review of literature". Blood Research. 51 (1): 23–30. doi:10.5045/br.2016.51.1.23. PMC 4828524. PMID 27104188.
- Bibliography
- Greer JP, Foerster J, and Lukens JN, "Wintrobe's Clinical Hematology", Lippincott Williams & Wilkins 11th ed., 2003.
- Hoffman R, Benz E, Shattil S, Furie B, Cohen H, "Hematology: Basic Principles and Practice", Churchill Livingstone, 4th ed, 2004.
- Hoffbrand AV, Catovsky D, and Tuddenham E, "Postgraduate Haematology", Blackwell, 5th ed., 2005.
- Hoffbrand AV, Moss PAH, and Pettit JE, "Essential Haematology", Blackwell, 5th ed., 2006.
