Copper extraction
Copper extraction refers to the methods used to obtain copper from its ores. The conversion of copper consists of a series of physical and electrochemical processes. Methods have evolved and vary with country depending on the ore source, local environmental regulations, and other factors.


As in all mining operations, the ore must usually be beneficiated (concentrated). The processing techniques depend on the nature of the ore. If the ore is primarily sulfide copper minerals (such as chalcopyrite), the ore is crushed and ground to liberate the valuable minerals from the waste ('gangue') minerals. It is then concentrated using mineral flotation. The concentrate is typically then sold to distant smelters, although some large mines have smelters located nearby. Such colocation of mines and smelters was more typical in the 19th and early 20th centuries, when smaller smelters could be economic. The sulfide concentrates are typically smelted in such furnaces as the Outokumpu or Inco flash furnace or the ISASMELT furnace to produce matte, which must be converted and refined to produce anode copper. Finally, the final refining process is electrolysis. For economic and environmental reasons, many of the byproducts of extraction are reclaimed. Sulfur dioxide gas, for example, is captured and turned into sulfuric acid — which can then be used in the extraction process or sold for such purposes as fertiliser manufacture.
Oxidised copper ores can be treated by hydrometallurgical extraction.
History

The earliest evidence of cold-hammering of native copper comes from the excavation at Çayönü Tepesi in eastern Anatolia, which dates between 7200 to 6600 BCE.[1] Among the various items considered to be votive or amulets there was one that looked like a fishhook and one like an awl. Another find, at Shanidar Cave in Mergasur, Iraq, contained copper beads, dates to 8,700 BCE.[2]
One of the world's oldest known copper mines, as opposed to usage of surface deposits, is at Timna Valley, Israel, since the fourth millennium BC, with surface deposit usage since the sixth to fifth millennium.[3][4]
The Pločnik archaeological site in southeastern Europe (Serbia) contains the oldest securely dated evidence of copper making at high temperature, from 5,000 BCE. The find in June 2010 extends for additional 500 years the earlier record of copper smelting from Rudna Glava (Serbia), dated to 5th millennium BCE.[5]
Copper smelting technology gave rise to the Copper Age, aka Chalcolithic Age, and then the Bronze Age. The Bronze Age would not have been possible without humans developing smelting technology.
Concentration
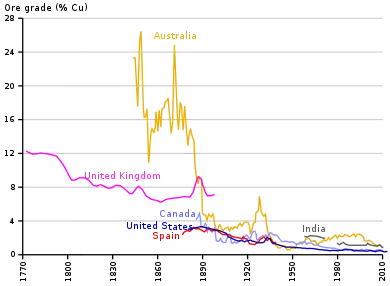
Most copper ores contain only a small percentage of copper. The remainder of the ore consists of gangue of no commercial value. Gangue from copper mining typically contains silicate minerals and oxides. In some cases these tailings have been retreated as the technology for recovering copper has improved. The average grade of copper ores in the 21st century is below 0.6% copper, with a proportion of economic ore minerals (including copper) being less than 2% of the total volume of the ore rock. A key objective in the metallurgical treatment of any ore is the separation of ore minerals from gangue minerals within the rock.
The first stage of any process within a metallurgical treatment circuit is accurate grinding or comminution, where the rock is crushed to produce small particles (<100 μm) consisting of individual mineral phases. These particles are then separated to remove gangue (rocks residues), thereafter followed by a process of physical liberation of the ore minerals from the rock. The process of liberation of copper ores depends upon whether they are oxide or sulfide ores.[6]
Subsequent steps depend on the nature of the ore containing the copper and what will be extracted. For oxide ores, a hydrometallurgical liberation process is normally undertaken, which uses the soluble nature of the ore minerals to the advantage of the metallurgical treatment plant. For sulfide ores, both secondary (supergene) and primary (hypogene), froth flotation is used to physically separate ore from gangue. For special native copper bearing ore bodies or sections of ore bodies rich in supergene native copper, this mineral can be recovered by a simple gravity circuit.
Froth flotation

The modern froth flotation process was independently invented the early 1900s in Australia by C.V Potter and around the same time by G. D. Delprat.[7]

All primary sulfide ores of copper sulfides, and most concentrates of secondary copper sulfides (being chalcocite), are subjected to smelting. Some vat leach or pressure leach processes exist to solubilise chalcocite concentrates and produce copper cathode from the resulting leachate solution, but this is a minor part of the market.
Carbonate concentrates are a relatively minor product produced from copper cementation plants, typically as the end-stage of a heap-leach operation. Such carbonate concentrates can be treated by a solvent extraction and electrowinning (SX-EW) plant or smelted.
The copper ore is crushed and ground to a size such that an acceptably high degree of liberation has occurred between the copper sulfide ore minerals and the gangue minerals. The ore is then wetted, suspended in a slurry, and mixed with xanthates or other reagents, which render the sulfide particles hydrophobic. Typical reagents include potassium ethylxanthate and sodium ethylxanthate, but dithiophosphates and dithiocarbamates are also used.
The treated ore is introduced to a water-filled aeration tank containing surfactant such as methylisobutyl carbinol (MIBC). Air is constantly forced through the slurry and the air bubbles attach to the hydrophobic copper sulfide particles, which are conducted to the surface, where they form a froth and are skimmed off. These skimmings are generally subjected to a cleaner-scavenger cell to remove excess silicates and to remove other sulfide minerals that can deleteriously impact the concentrate quality (typically, galena), and the final concentrate sent for smelting. The rock which has not floated off in the flotation cell is either discarded as tailings or further processed to extract other metals such as lead (from galena) and zinc (from sphalerite), should they exist. To improve the process efficiency, lime is used to raise the pH of the water bath, causing the collector to ionize more and to preferentially bond to chalcopyrite (CuFeS2) and avoid the pyrite (FeS2). Iron exists in both primary zone minerals. Copper ores containing chalcopyrite can be concentrated to produce a concentrate with between 20% and 30% copper-in-concentrate (usually 27–29% copper); the remainder of the concentrate is iron and sulfur in the chalcopyrite, and unwanted impurities such as silicate gangue minerals or other sulfide minerals, typically minor amounts of pyrite, sphalerite or galena. Chalcocite concentrates typically grade between 37% and 40% copper-in-concentrate, as chalcocite has no iron within the mineral.
Hydrometallurgical extraction
Sulfide ores
Secondary sulfides – those formed by supergene secondary enrichment – are resistant (refractory) to sulfuric leaching. These ores are a mixture of copper carbonate, sulfate, phosphate, and oxide minerals and secondary sulfide minerals, dominantly chalcocite but other minerals such as digenite can be important in some deposits.
Supergene ores rich in sulfides may be concentrated using froth flotation. A typical concentrate of chalcocite can grade between 37% and 40% copper in sulfide, making them relatively cheap to smelt compared to chalcopyrite concentrates.
Some supergene sulfide deposits can be leached using a bacterial oxidation heap leach process to oxidize the sulfides to sulfuric acid, which also allows for simultaneous leaching with sulfuric acid to produce a copper sulfate solution. As with oxide ores, solvent extraction and electrowinning technologies are used to recover the copper from the pregnant leach solution.
Supergene sulfide ores rich in native copper minerals are refractory to treatment with sulfuric acid leaching on all practicable time scales, and the dense metal particles do not react with froth flotation media. Typically, if native copper is a minor part of a supergene profile it will not be recovered and will report to the tailings. When rich enough, native copper ore bodies may be treated to recover the contained copper via a gravity separation circuit where the density of the metal is used to liberate it from the lighter silicate minerals. Often, the nature of the gangue is important, as clay-rich native copper ores prove difficult to liberate.
Oxide ores
Oxidised copper ore bodies may be treated via several processes, with hydrometallurgical processes used to treat oxide ores dominated by copper carbonate minerals such as azurite and malachite, and other soluble minerals such as silicates like chrysocolla, or sulfates such as atacamite and so on.
Such oxide ores are usually leached by sulfuric acid, usually in a heap leaching or dump leaching process to liberate the copper minerals into a solution of sulfuric acid laden with copper sulfate in solution. The copper sulfate solution (the pregnant leach solution) is then stripped of copper via a solvent extraction and electrowinning (SX-EW) plant, with the barred (denuded) sulfuric acid recycled back on to the heaps. Alternatively, the copper can be precipitated out of the pregnant solution by contacting it with scrap iron; a process called cementation. Cement copper is normally less pure than SX-EW copper. Commonly sulfuric acid is used as a leachant for copper oxide, although it is possible to use water, particularly for ores rich in ultra-soluble sulfate minerals.
In general, froth flotation is not used to concentrate copper oxide ores, as oxide minerals are not responsive to the froth flotation chemicals or process (i.e.; they do not bind to the kerosene-based chemicals). Copper oxide ores have occasionally been treated via froth flotation via sulfidation of the oxide minerals with certain chemicals which react with the oxide mineral particles to produce a thin rime of sulfide (usually chalcocite), which can then be activated by the froth flotation plant.
Sulfide smelting
Until the latter half of the 20th century, smelting sulfide ores was almost the sole means of producing copper metal from mined ores (primary copper production). Davenport, et al, noted in 2002 that even then 80% of global primary copper production was from copper–iron–sulfur minerals and that the vast majority of these were treated by smelting.[8]
Copper was initially recovered from sulfide ores by directly smelting the ore in a furnace.[9] The smelters were initially located near the mines to minimize the cost of transport. This avoided the prohibitive costs of transporting the waste minerals and the sulfur and iron present in the copper-containing minerals. However, as the concentration of copper in the ore bodies decreased, the energy costs of smelting the whole ore also became prohibitive, and it became necessary to concentrate the ores first.
Initial concentration techniques included hand-sorting[10] and gravity concentration. They resulted in high losses of copper. Consequently, the development of the froth flotation process was a major step forward in mineral processing.[11] It made possible the development of the giant Bingham Canyon mine in Utah.[12]
In the twentieth century, most ores were concentrated before smelting. Smelting was initially undertaken using sinter plants and blast furnaces,[13] or with roasters and reverberatory furnaces.[14] Roasting and reverberatory furnace smelting dominated primary copper production until the 1960s.[8]
Roasting
The roasting process is generally undertaken in combination with reverberatory furnaces. In the roaster, the copper concentrate is partially oxidised to produce "calcine" and sulfur dioxide gas. The stoichiometry of the reaction which occurs is:
- 2 CuFeS2 + 3 O2 → 2 FeO + 2 CuS + 2 SO2
Roasting generally leaves more sulfur in the calcined product (15% in the case of the roaster at Mount Isa Mines[15]) than a sinter plant leaves in the sintered product (about 7% in the case of the Electrolytic Refining and Smelting smelter[16]).
As of 2005, roasting is no longer common in copper concentrate treatment, because its combination with reverberatory furnaces is not energy efficient and the SO2 concentration in the roaster offgas is too dilute for cost-effective capture.[8] Direct smelting is now favored, e.g. using the following smelting technologies: flash smelting, Isasmelt, Noranda, Mitsubishi or El Teniente furnaces.[8]
Smelting
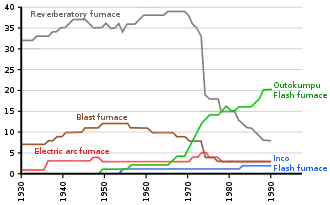
The initial melting of the material to be smelted is usually referred to as the smelting or matte smelting stage. It can be undertaken in a variety of furnaces, including the largely obsolete blast furnaces and reverberatory furnaces, as well as flash furnaces, Isasmelt furnaces, etc.[8] The product of this smelting stage is a mixture of copper, iron and sulfur that is enriched in copper, and which is called matte or copper matte.[8] The term matte grade is normally used to refer to the copper content of the matte.[17]
The purpose of the matte smelting stage is to eliminate as much of the unwanted iron, sulfur and gangue minerals (such as silica, magnesia, alumina and limestone) as possible, while minimizing the loss of copper.[17] This is achieved by reacting iron sulfides with oxygen (in air or oxygen enriched air) to produce iron oxides (mainly as FeO, but with some magnetite (Fe3O4)) and sulfur dioxide.[17]
Copper sulfide and iron oxide can mix, but when sufficient silica is added, a separate slag layer is formed.[18] Adding silica also reduces the melting point (or, more properly, the liquidus temperature) of the slag, meaning that the smelting process can be operated at a lower temperature.[18]
The slag forming reaction is:
- FeO + SiO2 → FeO.SiO2[17]
Slag is less dense than matte, so it forms a layer that floats on top of the matte.[19]
Copper can be lost from the matte in three ways: as cuprous oxide (Cu2O) dissolved in the slag,[20] as sulfide copper dissolved in the slag[21] or as tiny droplets (or prills) of matte suspended in the slag.[22][23]
The amount of copper lost as oxide copper increases as the oxygen potential of the slag increases.[23] The oxygen potential generally increases as the copper content of the matte is increased.[24] Thus the loss of copper as oxide increases as the copper content of the matte increases.[25]
On the other hand, the solubility of sulfidic copper in slag decreases as the copper content of the matte increases beyond about 40%.[21] Nagamori calculated that more than half the copper dissolved in slags from mattes containing less than 50% copper is sulfidic copper. Above this figure, oxidic copper begins to dominate.[21]
The loss of copper as prills suspended in the slag depends on the size of the prills, the viscosity of the slag and the settling time available.[26] Rosenqvist suggested that about half the copper losses to slag were due to suspended prills.[26]
The mass of slag generated in the smelting stage depends on the iron content of the material fed into the smelting furnace and the target matte grade. The greater the iron content of the feed, the more iron that will need to be rejected to the slag for a given matte grade. Similarly, increasing the target matte grade requires the rejection of more iron and an increase in the slag volume.
Thus, the two factors that most affect the loss of copper to slag in the smelting stage are:
- matte grade
- mass of slag.[18]
This means that there is a practical limit on how high the matte grade can be if the loss of copper to slag is to be minimized. Therefore, further stages of processing (converting and fire refining) are required.
The following subsections briefly describe some of the processes used in matte smelting.
Reverberatory furnace smelting
Reverberatory furnaces are long furnaces can treat wet, dry or roasted concentrate.[8] Most of the reverberatory furnaces used in the latter years treated roasted concentrate because putting dry feed materials into the reverberatory furnace is more energy efficient, and because the elimination of some of the sulfur in the roaster results in higher matte grades.[8]
The reverberatory furnace feed is added to the furnace through feed holes along the sides of the furnace.[8] Additional silica is normally added to help form the slag. The furnace is fired with burners using pulverized coal, fuel oil or natural gas[27] and the solid charge is melted.
Reverberatory furnaces can additionally be fed with molten slag from the later converting stage to recover the contained copper and other materials with a high copper content.[27]
Because the reverberatory furnace bath is quiescent, very little oxidation of the feed occurs (and thus very little sulfur is eliminated from the concentrate). It is essentially a melting process.[26] Consequently, wet-charged reverberatory furnaces have less copper in their matte product than calcine-charged furnaces, and they also have lower copper losses to slag.[27] Gill quotes a copper in slag value of 0.23% for a wet-charged reverberatory furnace vs 0.37% for a calcine-charged furnace.[27]
In the case of calcine-charged furnaces, a significant portion of the sulfur has been eliminated during the roasting stage, and the calcine consists of a mixture of copper and iron oxides and sulfides. The reverberatory furnace acts to allow these species to approach chemical equilibrium at the furnace operating temperature (approximately 1600 °C at the burner end of the furnace and about 1200 °C at the flue end;[28] the matte is about 1100 °C and the slag is about 1195 °C[27]). In this equilibration process, oxygen associated with copper compounds exchanges with sulfur associated with iron compounds, increasing the iron oxide content of the furnace, and the iron oxides interact with silica and other oxide materials to form the slag.[27]
The main equilibration reaction is:
- Cu2O + FeS = Cu2S + FeO[27]
The slag and the matte form distinct layers that can be removed from the furnace as separate streams. The slag layer is periodically allowed to flow through a hole in the wall of the furnace above the height of the matte layer. The matte is removed by draining it through a hole into ladles for it to be carried by crane to the converters.[27] This draining process is known as tapping the furnace.[27] The matte taphole is normally a hole through a water-cooled copper block that prevents erosion of the refractory bricks lining the furnace. When the removal of the matte or slag is complete, the hole is normally plugged with clay, which is removed when the furnace is ready to be tapped again.
Reverberatory furnaces were often used to treat molten converter slag to recover contained copper.[27] This would be poured into the furnaces from ladles carried by cranes. However, the converter slag is high in magnetite[29] and some of this magnetite would precipitate from the converter slag (due to its higher melting point), forming an accretion on the hearth of the reverberatory furnace and necessitating shut downs of the furnace to remove the accretion.[29] This accretion formation limits the quantity of converter slag that can be treated in a reverberatory furnace.[29]
While reverberatory furnaces have very low copper losses to slag, they are not very energy-efficient and the low concentrations of sulfur dioxide in their offgases make its capture uneconomic.[8] Consequently, smelter operators devoted a lot of money in the 1970s and 1980s to developing new, more efficient copper smelting processes.[30] In addition, flash smelting technologies had been developed in earlier years and began to replace reverberatory furnaces. By 2002, 20 of the 30 reverberatory furnaces still operating in 1994 had been shut down.[8]
Flash furnace smelting
In flash smelting, the concentrate is dispersed in an air or oxygen stream and the smelting reactions are largely completed while the mineral particles are still in flight.[30] The reacted particles then settle in a bath at the bottom of the furnace, where they behave as does calcine in a reverberatory furnace.[31] A slag layer forms on top of the matte layer, and they can separately be tapped from the furnace.[31]
Converting

The matte, which is produced in the smelter, contains 30–70% copper (depending on the process used and the operating philosophy of the smelter), primarily as copper sulfide, as well as iron sulfide. The sulfur is removed at high temperature as sulfur dioxide by blowing air through molten matte:
- 2 CuS + 3 O2 → 2 CuO + 2 SO2
- CuS + O2 → Cu + SO2
In a parallel reaction the iron sulfide is converted to slag:
- 2 FeS + 3 O2 → 2 FeO + 2 SO2
- 2 FeO + SiO2 → Fe2SiO4
The purity of this product is 98%, it is known as blister because of the broken surface created by the escape of sulfur dioxide gas as blister copper pigs or ingots are cooled. By-products generated in the process are sulfur dioxide and slag. The sulfur dioxide is captured for use in earlier leaching processes.
Fire refining
The blister copper is put into an anode furnace, a furnace that refines the blister copper to anode-grade copper in two stages by removing most of the remaining sulfur and iron, and then removing oxygen introduced during the first stage. This second stage, often referred to as poling is done by blowing natural gas, or some other reducing agent, through the molten copper oxide. When this flame burns green, indicating the copper oxidation spectrum, the oxygen has mostly been burned off. This creates copper at about 99% pure.
Electrorefining
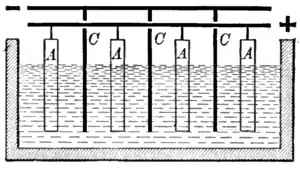
The copper is refined by electrolysis. The anodes cast from processed blister copper are placed into an aqueous solution of 3–4% copper sulfate and 10–16% sulfuric acid. Cathodes are thin rolled sheets of highly pure copper or, more commonly these days, reusable stainless steel starting sheets (as in the IsaKidd process).[32] A potential of only 0.2–0.4 volts is required for the process to commence. In industrial plants current densities up to 420 A/m2 are possible.[33] At the anode, copper and less noble metals dissolve. More noble metals such as silver, gold, selenium, and tellurium settle to the bottom of the cell as anode slime, which forms a salable byproduct. Copper(II) ions migrate through the electrolyte to the cathode. At the cathode, copper metal plates out, but less noble constituents such as arsenic and zinc remain in solution unless a higher voltage is used.[34] The reactions are:
At the anode: Cu(s) → Cu2+(aq) + 2e−
At the cathode: Cu2+(aq) + 2e− → Cu(s)
Concentrate and copper marketing
Copper concentrates produced by mines are sold to smelters and refiners who treat the ore and refine the copper and charge for this service via treatment charges (TCs) and refining charges (RCs). The global copper concentrate market [35]was valued at US$ 81 Billion in 2019 and is projected to reach US$ 93 Billion by 2027 expanding at a CAGR of 2.5%. The TCs are charged in US$ per tonne of concentrate treated and RCs are charged in cents per pound treated, denominated in US dollars, with benchmark prices set annually by major Japanese smelters. The customer in this case can be a smelter, who on-sells blister copper ingots to a refiner, or a smelter-refiner which is vertically integrated.
One prevalent form of copper concentrate contains gold and silver, like the one produced by Bougainville Copper Limited from the Panguna mine from the early 1970s to the late 1980s.[36]
The typical contract for a miner is denominated against the London Metal Exchange price, minus the TC-RCs and any applicable penalties or credits. Penalties may be assessed against copper concentrates according to the level of deleterious elements such as arsenic, bismuth, lead or tungsten. Because a large portion of copper sulfide ore bodies contain silver or gold in appreciable amounts, a credit can be paid to the miner for these metals if their concentration within the concentrate is above a certain amount. Usually the refiner or smelter charges the miner a fee based on the concentration; a typical contract will specify that a credit is due for every ounce of the metal in the concentrate above a certain concentration; below that, if it is recovered, the smelter will keep the metal and sell it to defray costs.
Copper concentrate is traded either via spot contracts or under long term contracts as an intermediate product in its own right. Often the smelter sells the copper metal itself on behalf of the miner. The miner is paid the price at the time that the smelter-refiner makes the sale, not at the price on the date of delivery of the concentrate. Under a Quotational Pricing system, the price is agreed to be at a fixed date in the future, typically 90 days from time of delivery to the smelter.
A-grade copper cathode is of 99.99% copper in sheets that are 1 cm thick, and approximately 1 meter square weighing approximately 200 pounds. It is a true commodity, deliverable to and tradeable upon the metal exchanges in New York City (COMEX), London (London Metals Exchange) and Shanghai (Shanghai Futures Exchange). Often copper cathode is traded upon the exchanges indirectly via warrants, options, or swap contracts such that the majority of copper is traded upon the LME/COMEX/SFE but delivery is achieved directly, logistically moving the physical copper, and transferring the copper sheet from the physical warehouses themselves.
The chemical specification for electrolytic grade copper is ASTM B 115-00 (a standard that specifies the purity and maximum electrical resistivity of the product).
See also
References
- Robert J. Braidwood; Halet Çambel; Charles L. Redman; Patty Jo Watson (1971). "Beginnings of Village-Farming Communities in Southeastern Turkey". Proc Natl Acad Sci U S A. 68 (6): 1236–1240. Bibcode:1971PNAS...68.1236B. doi:10.1073/pnas.68.6.1236. PMC 389161. PMID 16591930.
- R.S. Solecki; R.L. Solecki; A.P. Agelarakis (2004). The Proto-neolithic Cemetery in Shanidar Cave. Texas A&M University Press. p. 53. ISBN 9781585442720.
- Ian Shaw (2002-05-06). A Dictionary of Archaeology. John Wiley & Sons. pp. 582–583. ISBN 9780631235835.
- PJ King; LE Stager (2001-01-01). Life In Biblical Israel. Westminster John Knox Press. p. 165. ISBN 9780664221485.
timna millennium.
- "Serbian site may have hosted first copper makers". ScienceNews. July 17, 2010.
- Adalbert Lossin "Copper" in Ullmann's Encyclopedia of Industrial Chemistry 2005, Wiley-VCH, Weinheim. doi:10.1002/14356007.a07_471
- "Historical Note". Minerals Separation Ltd. Retrieved 2007-12-30.
- W G Davenport, M King, M Schlesinger and A K Biswas, Extractive Metallurgy of Copper, Fourth Edition (Elsevier Science Limited: Kidlington, Oxford, England, 2002).
- Robert Raymond, Out of the Fiery Furnace, The MacMillan Company of Australia Pty Ltd, Melbourne, 1984.
- C B Gill, Non-ferrous Extractive Metallurgy (John Wiley & Sons, New York, 1980) p. 32
- Robert Raymond (1984) Out of the Fiery Furnace, The MacMillan Company of Australia Pty Ltd, Melbourne, pp. 233–235, ISBN 027100441X.
- "BP Minerals completes $400 million modernization at Bingham Canyon," Mining Engineering, November 1988, 1017–1020.
- S A Bradford (1989) "The historical development of copper smelting in British Columbia," in: All that Glitters: Readings in Historical Metallurgy, Ed. Michael L. Wayman, The Canadian Institute of Mining and Metallurgy: Montreal, pp. 162–165, ISBN 0919086241.
- E Kossatz and P J Mackey (1989) "The first copper smelter in Canada," in: All that Glitters: Readings in Historical Metallurgy, Ed. Michael L. Wayman, The Canadian Institute of Mining and Metallurgy: Montreal, pp. 160–161, ISBN 0919086241.
- B V Borgelt, G E Casley and J Pritchard (1974) "Fluid bed roasting at Mount Isa," The Aus.I.M.M. North West Queensland Branch, Regional Meeting, August 1974. The Australasian Institute of Mining and Metallurgy: Melbourne, pp. 123–130.
- P J Wand (1980) "Copper smelting at Electrolytic Refining and Smelting Company of Australia Ltd., Port Kembla, N.S.W.", in: Mining and Metallurgical Practices in Australasia: The Sir Maurice Mawby Memorial Volume, Ed J T Woodcock. The Australasian Institute of Mining and Metallurgy: Melbourne. pp. 335–340.
- W G Davenport, M King, M Schlesinger and A K Biswas, Extractive Metallurgy of Copper, Fourth Edition (Elsevier Science Limited: Kidlington, Oxford, England, 2002), 57–72.
- P C Hayes, Process Principles in Minerals and Materials Production (Hayes Publishing Company: Brisbane, 1993), 173–179.
- C B Gill, Non-ferrous Extractive Metallurgy (John Wiley & Sons, New York, 1980) p. 19
- R Altman and H H Kellogg, "Solubility of copper in silica-saturated iron silicate slag," Transactions of the Institution of Mining and Metallurgy (Section C: Mineral Processing and Extractive Metallurgy), 81, September 1972, C163–C175.
- M Nagamori (1974). "Metal loss to slag: Part I. Sulfidic and oxidic dissolution of copper in fayalite slag from low grade matte". Metallurgical Transactions. 5 (3): 531–538. Bibcode:1974MT......5..531N. doi:10.1007/BF02644646. S2CID 135507603.
- A Yazawa and S Nakazawa, "Evaluation of non-equilibrium minor components in pyrometallurgy," in: EPD Congress 1998, Ed. B Mishra (The Minerals, Metals and Materials Society: Warrendale, Pennsylvania, 1998), 641–655.
- B J Elliott, J B See, and W J Rankin, "Effect of slag composition on copper losses to silica-saturated iron silicate slags," Transactions of the Institution of Mining and Metallurgy (Section C: Mineral Processing and Extractive Metallurgy), September 1978, C–C211.
- J Matousek (1993). "Oxygen potentials of copper smelting slags". Canadian Metallurgical Quarterly. 32 (2): 97–101. doi:10.1179/cmq.1993.32.2.97.
- P J Mackey (2011). "The Physical Chemistry of Copper Smelting Slags and Copper Losses at the Paipote SmelterPart 2 – Characterisation of industrial slags". Canadian Metallurgical Quarterly. 50 (4): 330–340. doi:10.1179/000844311X13112418194806. S2CID 137350753.
- T Rosenqvist, T (2004) Principles of Extractive Metallurgy, Second Edition, Tapir Academic Press: Trondheim, p. 331, ISBN 8251919223.
- C B Gill, Non-ferrous Extractive Metallurgy (John Wiley & Sons, New York, 1980) pp. 29–35
- C B Gill, Non-ferrous Extractive Metallurgy (John Wiley & Sons, New York, 1980) p. 23
- G E Casley, J Middlin and D White, "Recent developments in reverberatory furnace and converter practice at the Mount Isa Mines copper smelter," in: Extractive Metallurgy of Copper, Volume 1, (The Metallurgical Society: Warrendale, Pennsylvania, 1976), 117–138.
- P J Mackey and P Tarassoff, "New and emerging technologies in sulphide [sic] smelting," in: Advances in Sulfide Smelting Volume 2: Technology and Practice, Eds H Y Sohn, D B George and A D Zunkel (The Metallurgical Society of the American Institute of Mining, Metallurgical and Petroleum Engineers: Warrendale, Pennsylvania, 1983), 399–426.
- W G Davenport, M King, M Schlesinger and A K Biswas, Extractive Metallurgy of Copper, Fourth Edition (Elsevier Science Limited: Kidlington, Oxford, England, 2002), 73–102.
- T Robinson, "Electrolytic refining," in: Extractive Metallurgy of Copper, Fourth Edition, Eds W G Davenport, M King, M Schlesinger and A K Biswas (Elsevier Science Limited: Kidlington, Oxford, England, 2002) 265–288.
- A. Filzwieser, M.B. Hanel, I. Filzwieser, S. Wallner (May 13, 2019). "FAQS WITH REGARDS TO OPERATING A CU ER TANKHOUSE ABOVE 400 A/m2" (PDF).CS1 maint: multiple names: authors list (link)
- Samans, Carl H. Engineering Metals and their Alloys MacMillan 1949
- Reports, Growth Market. "Copper Concentrate Market - Global Industry Analysis, Size, Share, Growth, Trends, and Forecast | Growth Market Reports". growthmarketreports.com. Retrieved 2020-08-24.
- "About the Company". Archived from the original on 23 September 2015. Retrieved 24 August 2015.
Bibliography
- Gill, C. B. (1980) Nonferrous Extractive Metallurgy, John Wiley and Sons: New York, ISBN 0471059803
External links
| Wikisource has the text of the 1879 American Cyclopædia article Copper Smelting. |
- The Copper Development Association's copper production page.
- Copper Processing, from an online text on metals, Illinois Sustainable Technology Center, University of Illinois, archived in May 2017
- National Pollutant Inventory – Copper and copper compounds fact sheet
- University of Pittsburgh School of Engineering Chemical and Petroleum Engineering Department, Froth Flotation Lab notes.
- Copper Mine, college course syllabus, emphasis on chemistry, last updated 2000
