Fermium
Fermium is a synthetic element with the symbol Fm and atomic number 100. It is an actinide and the heaviest element that can be formed by neutron bombardment of lighter elements, and hence the last element that can be prepared in macroscopic quantities, although pure fermium metal has not yet been prepared.[3] A total of 19 isotopes are known, with 257Fm being the longest-lived with a half-life of 100.5 days.
| Fermium | ||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pronunciation | /ˈfɜːrmiəm/ | |||||||||||||||||||||||||||||||||
| Mass number | [257] | |||||||||||||||||||||||||||||||||
| Fermium in the periodic table | ||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||
| Atomic number (Z) | 100 | |||||||||||||||||||||||||||||||||
| Group | n/a | |||||||||||||||||||||||||||||||||
| Period | period 7 | |||||||||||||||||||||||||||||||||
| Block | f-block | |||||||||||||||||||||||||||||||||
| Electron configuration | [Rn] 5f12 7s2 | |||||||||||||||||||||||||||||||||
| Electrons per shell | 2, 8, 18, 32, 30, 8, 2 | |||||||||||||||||||||||||||||||||
| Physical properties | ||||||||||||||||||||||||||||||||||
| Phase at STP | solid (predicted) | |||||||||||||||||||||||||||||||||
| Melting point | 1800 K (1527 °C, 2781 °F) (predicted) | |||||||||||||||||||||||||||||||||
| Density (near r.t.) | 9.7(1) g/cm3 (predicted)[1] | |||||||||||||||||||||||||||||||||
| Atomic properties | ||||||||||||||||||||||||||||||||||
| Oxidation states | +2, +3 | |||||||||||||||||||||||||||||||||
| Electronegativity | Pauling scale: 1.3 | |||||||||||||||||||||||||||||||||
| Ionization energies |
| |||||||||||||||||||||||||||||||||
| Other properties | ||||||||||||||||||||||||||||||||||
| Natural occurrence | synthetic | |||||||||||||||||||||||||||||||||
| Crystal structure | face-centered cubic (fcc) (predicted)[1] | |||||||||||||||||||||||||||||||||
| CAS Number | 7440-72-4 | |||||||||||||||||||||||||||||||||
| History | ||||||||||||||||||||||||||||||||||
| Naming | after Enrico Fermi | |||||||||||||||||||||||||||||||||
| Discovery | Lawrence Berkeley National Laboratory (1952) | |||||||||||||||||||||||||||||||||
| Main isotopes of fermium | ||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||
It was discovered in the debris of the first hydrogen bomb explosion in 1952, and named after Enrico Fermi, one of the pioneers of nuclear physics. Its chemistry is typical for the late actinides, with a preponderance of the +3 oxidation state but also an accessible +2 oxidation state. Owing to the small amounts of produced fermium and all of its isotopes having relatively short half-lives, there are currently no uses for it outside basic scientific research.
Discovery



Fermium was first discovered in the fallout from the 'Ivy Mike' nuclear test (1 November 1952), the first successful test of a hydrogen bomb.[4][5][6] Initial examination of the debris from the explosion had shown the production of a new isotope of plutonium, 244
94Pu
: this could only have formed by the absorption of six neutrons by a uranium-238 nucleus followed by two β− decays. At the time, the absorption of neutrons by a heavy nucleus was thought to be a rare process, but the identification of 244
94Pu
raised the possibility that still more neutrons could have been absorbed by the uranium nuclei, leading to new elements.[6]
Element 99 (einsteinium) was quickly discovered on filter papers which had been flown through the cloud from the explosion (the same sampling technique that had been used to discover 244
94Pu
).[6] It was then identified in December 1952 by Albert Ghiorso and co-workers at the University of California at Berkeley.[4][5][6] They discovered the isotope 253Es (half-life 20.5 days) that was made by the capture of 15 neutrons by uranium-238 nuclei – which then underwent seven successive beta decays:
(1)

Some 238U atoms, however, could capture another amount of neutrons (most likely, 16 or 17).
The discovery of fermium (Z = 100) required more material, as the yield was expected to be at least an order of magnitude lower than that of element 99, and so contaminated coral from the Enewetak atoll (where the test had taken place) was shipped to the University of California Radiation Laboratory in Berkeley, California, for processing and analysis. About two months after the test, a new component was isolated emitting high-energy α-particles (7.1 MeV) with a half-life of about a day. With such a short half-life, it could only arise from the β− decay of an isotope of einsteinium, and so had to be an isotope of the new element 100: it was quickly identified as 255Fm (t = 20.07(7) hours).[6]
The discovery of the new elements, and the new data on neutron capture, was initially kept secret on the orders of the U.S. military until 1955 due to Cold War tensions.[6][7][8] Nevertheless, the Berkeley team was able to prepare elements 99 and 100 by civilian means, through the neutron bombardment of plutonium-239, and published this work in 1954 with the disclaimer that it was not the first studies that had been carried out on the elements.[9][10] The "Ivy Mike" studies were declassified and published in 1955.[7]
The Berkeley team had been worried that another group might discover lighter isotopes of element 100 through ion-bombardment techniques before they could publish their classified research,[6] and this proved to be the case. A group at the Nobel Institute for Physics in Stockholm independently discovered the element, producing an isotope later confirmed to be 250Fm (t1/2 = 30 minutes) by bombarding a 238
92U
target with oxygen-16 ions, and published their work in May 1954.[11] Nevertheless, the priority of the Berkeley team was generally recognized, and with it the prerogative to name the new element in honour of the recently deceased Enrico Fermi, the developer of the first artificial self-sustained nuclear reactor.
Isotopes
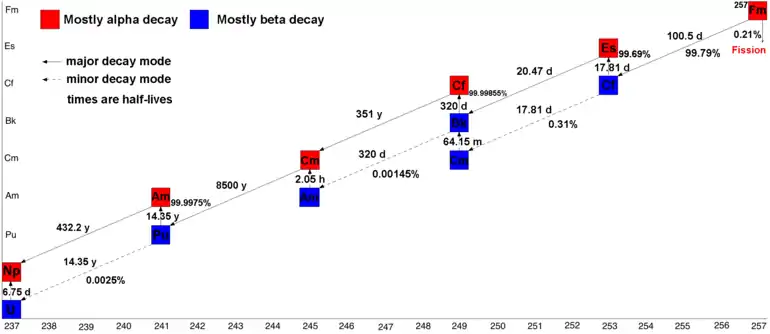
There are 20 isotopes of fermium listed in NUBASE 2016,[12] with atomic weights of 241 to 260,[Note 1] of which 257Fm is the longest-lived with a half-life of 100.5 days. 253Fm has a half-life of 3 days, while 251Fm of 5.3 h, 252Fm of 25.4 h, 254Fm of 3.2 h, 255Fm of 20.1 h, and 256Fm of 2.6 hours. All the remaining ones have half-lives ranging from 30 minutes to less than a millisecond.[13] The neutron capture product of fermium-257, 258Fm, undergoes spontaneous fission with a half-life of just 370(14) microseconds; 259Fm and 260Fm are also unstable with respect to spontaneous fission (t1/2 = 1.5(3) s and 4 ms respectively).[13] This means that neutron capture cannot be used to create nuclides with a mass number greater than 257, unless carried out in a nuclear explosion. As 257Fm is an α-emitter, decaying to 253Cf, and no known fermium isotopes undergo beta minus decay to the next element, mendelevium, fermium is also the last element that can be prepared by a neutron-capture process.[3][14][15] Because of this impediment in forming heavier isotopes, these short-lived isotopes 258–260Fm constitute the so-called "fermium gap."[16]
Production
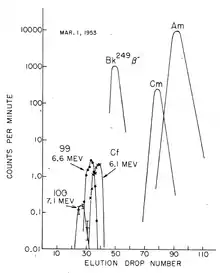
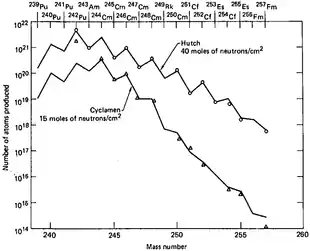
Fermium is produced by the bombardment of lighter actinides with neutrons in a nuclear reactor. Fermium-257 is the heaviest isotope that is obtained via neutron capture, and can only be produced in picogram quantities.[Note 2][17] The major source is the 85 MW High Flux Isotope Reactor (HFIR) at the Oak Ridge National Laboratory in Tennessee, USA, which is dedicated to the production of transcurium (Z > 96) elements.[18] Lower mass fermium isotopes are available in greater quantities, though these isotopes (254Fm and 255Fm) are comparatively short-lived. In a "typical processing campaign" at Oak Ridge, tens of grams of curium are irradiated to produce decigram quantities of californium, milligram quantities of berkelium and einsteinium, and picogram quantities of fermium.[19] However, nanogram[20] quantities of fermium can be prepared for specific experiments. The quantities of fermium produced in 20–200 kiloton thermonuclear explosions is believed to be of the order of milligrams, although it is mixed in with a huge quantity of debris; 4.0 picograms of 257Fm was recovered from 10 kilograms of debris from the "Hutch" test (16 July 1969).[21] The Hutch experiment produced an estimated total of 250 micrograms of 257Fm.
After production, the fermium must be separated from other actinides and from lanthanide fission products. This is usually achieved by ion-exchange chromatography, with the standard process using a cation exchanger such as Dowex 50 or TEVA eluted with a solution of ammonium α-hydroxyisobutyrate.[3][22] Smaller cations form more stable complexes with the α-hydroxyisobutyrate anion, and so are preferentially eluted from the column.[3] A rapid fractional crystallization method has also been described.[3][23]
Although the most stable isotope of fermium is 257Fm, with a half-life of 100.5 days, most studies are conducted on 255Fm (t1/2 = 20.07(7) hours), since this isotope can be easily isolated as required as the decay product of 255Es (t1/2 = 39.8(12) days).[3]
Synthesis in nuclear explosions
The analysis of the debris at the 10-megaton Ivy Mike nuclear test was a part of long-term project, one of the goals of which was studying the efficiency of production of transuranium elements in high-power nuclear explosions. The motivation for these experiments was as follows: synthesis of such elements from uranium requires multiple neutron capture. The probability of such events increases with the neutron flux, and nuclear explosions are the most powerful neutron sources, providing densities of the order 1023 neutrons/cm2 within a microsecond, i.e. about 1029 neutrons/(cm2·s). In comparison, the flux of the HFIR reactor is 5×1015 neutrons/(cm2·s). A dedicated laboratory was set up right at Enewetak Atoll for preliminary analysis of debris, as some isotopes could have decayed by the time the debris samples reached the U.S. The laboratory was receiving samples for analysis, as soon as possible, from airplanes equipped with paper filters which flew over the atoll after the tests. Whereas it was hoped to discover new chemical elements heavier than fermium, those were not found after a series of megaton explosions conducted between 1954 and 1956 at the atoll.[24]
The atmospheric results were supplemented by the underground test data accumulated in the 1960s at the Nevada Test Site, as it was hoped that powerful explosions conducted in confined space might result in improved yields and heavier isotopes. Apart from traditional uranium charges, combinations of uranium with americium and thorium have been tried, as well as a mixed plutonium-neptunium charge. They were less successful in terms of yield that was attributed to stronger losses of heavy isotopes due to enhanced fission rates in heavy-element charges. Isolation of the products was found to be rather problematic, as the explosions were spreading debris through melting and vaporizing rocks under the great depth of 300–600 meters, and drilling to such depth in order to extract the products was both slow and inefficient in terms of collected volumes.[24][25]
Among the nine underground tests, which were carried between 1962 and 1969 and codenamed Anacostia (5.2 kilotons, 1962), Kennebec (<5 kilotons, 1963), Par (38 kilotons, 1964), Barbel (<20 kilotons, 1964), Tweed (<20 kilotons, 1965), Cyclamen (13 kilotons, 1966), Kankakee (20-200 kilotons, 1966), Vulcan (25 kilotons, 1966) and Hutch (20-200 kilotons, 1969),[26] the last one was most powerful and had the highest yield of transuranium elements. In the dependence on the atomic mass number, the yield showed a saw-tooth behavior with the lower values for odd isotopes, due to their higher fission rates.[25] The major practical problem of the entire proposal, however, was collecting the radioactive debris dispersed by the powerful blast. Aircraft filters adsorbed only about 4×10−14 of the total amount and collection of tons of corals at Enewetak Atoll increased this fraction by only two orders of magnitude. Extraction of about 500 kilograms of underground rocks 60 days after the Hutch explosion recovered only about 10−7 of the total charge. The amount of transuranium elements in this 500-kg batch was only 30 times higher than in a 0.4 kg rock picked up 7 days after the test. This observation demonstrated the highly nonlinear dependence of the transuranium elements yield on the amount of retrieved radioactive rock.[27] In order to accelerate sample collection after explosion, shafts were drilled at the site not after but before the test, so that explosion would expel radioactive material from the epicenter, through the shafts, to collecting volumes near the surface. This method was tried in the Anacostia and Kennebec tests and instantly provided hundreds kilograms of material, but with actinide concentration 3 times lower than in samples obtained after drilling; whereas such method could have been efficient in scientific studies of short-lived isotopes, it could not improve the overall collection efficiency of the produced actinides.[28]
Although no new elements (apart from einsteinium and fermium) could be detected in the nuclear test debris, and the total yields of transuranium elements were disappointingly low, these tests did provide significantly higher amounts of rare heavy isotopes than previously available in laboratories. For example, 6×109 atoms of 257Fm could be recovered after the Hutch detonation. They were then used in the studies of thermal-neutron induced fission of 257Fm and in discovery of a new fermium isotope 258Fm. Also, the rare 250Cm isotope was synthesized in large quantities, which is very difficult to produce in nuclear reactors from its progenitor 249Cm – the half-life of 249Cm (64 minutes) is much too short for months-long reactor irradiations, but is very "long" on the explosion timescale.[29]
Natural occurrence
Because of the short half-life of all isotopes of fermium, any primordial fermium, that is fermium that could be present on the Earth during its formation, has decayed by now. Synthesis of fermium from naturally occurring actinides uranium and thorium in the Earth crust requires multiple neutron capture, which is an extremely unlikely event. Therefore, most fermium is produced on Earth in scientific laboratories, high-power nuclear reactors, or in nuclear weapons tests, and is present only within a few months from the time of the synthesis. The transuranic elements from americium to fermium did occur naturally in the natural nuclear fission reactor at Oklo, but no longer do so.[30]
Chemistry
The chemistry of fermium has only been studied in solution using tracer techniques, and no solid compounds have been prepared. Under normal conditions, fermium exists in solution as the Fm3+ ion, which has a hydration number of 16.9 and an acid dissociation constant of 1.6×10−4 (pKa = 3.8).[31][32] Fm3+ forms complexes with a wide variety of organic ligands with hard donor atoms such as oxygen, and these complexes are usually more stable than those of the preceding actinides.[3] It also forms anionic complexes with ligands such as chloride or nitrate and, again, these complexes appear to be more stable than those formed by einsteinium or californium.[33] It is believed that the bonding in the complexes of the later actinides is mostly ionic in character: the Fm3+ ion is expected to be smaller than the preceding An3+ ions because of the higher effective nuclear charge of fermium, and hence fermium would be expected to form shorter and stronger metal–ligand bonds.[3]
Fermium(III) can be fairly easily reduced to fermium(II),[34] for example with samarium(II) chloride, with which fermium(II) coprecipitates.[35][36] In the precipitate, the compound fermium(II) chloride (FmCl2) was produced, though it was not purified or studied in isolation.[37] The electrode potential has been estimated to be similar to that of the ytterbium(III)/(II) couple, or about −1.15 V with respect to the standard hydrogen electrode,[38] a value which agrees with theoretical calculations.[39] The Fm2+/Fm0 couple has an electrode potential of −2.37(10) V based on polarographic measurements.[40]
Toxicity
Although few people come in contact with fermium, the International Commission on Radiological Protection has set annual exposure limits for the two most stable isotopes. For fermium-253, the ingestion limit was set at 107 becquerels (1 Bq is equivalent to one decay per second), and the inhalation limit at 105 Bq; for fermium-257, at 105 Bq and 4000 Bq respectively.[41]
Notes and references
Notes
- The discovery of 260Fm is considered "unproven" in NUBASE 2003.[13]
- All isotopes of elements Z > 100 can only be produced by accelerator-based nuclear reactions with charged particles and can be obtained only in tracer quantities (e.g., 1 million atoms for Md (Z = 101) per hour of irradiation (see reference 1 below)).
References
- Fournier, Jean-Marc (1976). "Bonding and the electronic structure of the actinide metals". Journal of Physics and Chemistry of Solids. 37 (2): 235–244. Bibcode:1976JPCS...37..235F. doi:10.1016/0022-3697(76)90167-0.
- https://pubs.acs.org/doi/10.1021/jacs.8b09068
- Silva, Robert J. (2006). "Fermium, Mendelevium, Nobelium, and Lawrencium" (PDF). In Morss, Lester R.; Edelstein, Norman M.; Fuger, Jean (eds.). The Chemistry of the Actinide and Transactinide Elements. 3 (3rd ed.). Dordrecht: Springer. pp. 1621–1651. doi:10.1007/1-4020-3598-5_13. ISBN 978-1-4020-3555-5. Archived from the original (PDF) on 17 July 2010.
- "Einsteinium". Archived from the original on 26 October 2007. Retrieved 7 December 2007.
- Fermium – National Research Council Canada Archived 2010-12-25 at the Wayback Machine. Retrieved 2 December 2007
- Ghiorso, Albert (2003). "Einsteinium and Fermium". Chemical and Engineering News. 81 (36): 174–175. doi:10.1021/cen-v081n036.p174.
- Ghiorso, A.; Thompson, S.; Higgins, G.; Seaborg, Glenn T.; Studier, M.; Fields, P.; Fried, S.; Diamond, H.; et al. (1955). "New Elements Einsteinium and Fermium, Atomic Numbers 99 and 100". Phys. Rev. 99 (3): 1048–1049. Bibcode:1955PhRv...99.1048G. doi:10.1103/PhysRev.99.1048.
- Fields, P. R.; Studier, M. H.; Diamond, H.; Mech, J. F.; Inghram, M. G. Pyle, G. L.; Stevens, C. M.; Fried, S.; Manning, W. M. (Argonne National Laboratory, Lemont, Illinois); Ghiorso, A.; Thompson, S. G.; Higgins, G. H.; Seaborg, G. T. (University of California, Berkeley, California): "Transplutonium Elements in Thermonuclear Test Debris", in: Fields, P.; Studier, M.; Diamond, H.; Mech, J.; Inghram, M.; Pyle, G.; Stevens, C.; Fried, S.; Manning, W.; Ghiorso, A.; Thompson, S.; Higgins, G.; Seaborg, G. (1956). "Transplutonium Elements in Thermonuclear Test Debris". Physical Review. 102 (1): 180. Bibcode:1956PhRv..102..180F. doi:10.1103/PhysRev.102.180.
- Thompson, S. G.; Ghiorso, A.; Harvey, B. G.; Choppin, G. R. (1954). "Transcurium Isotopes Produced in the Neutron Irradiation of Plutonium". Physical Review. 93 (4): 908. Bibcode:1954PhRv...93..908T. doi:10.1103/PhysRev.93.908.
- Choppin, G. R.; Thompson, S. G.; Ghiorso, A.; Harvey, B. G. (1954). "Nuclear Properties of Some Isotopes of Californium, Elements 99 and 100". Physical Review. 94 (4): 1080–1081. Bibcode:1954PhRv...94.1080C. doi:10.1103/PhysRev.94.1080.
- Atterling, Hugo; Forsling, Wilhelm; Holm, Lennart W.; Melander, Lars; Åström, Björn (1954). "Element 100 Produced by Means of Cyclotron-Accelerated Oxygen Ions". Physical Review. 95 (2): 585–586. Bibcode:1954PhRv...95..585A. doi:10.1103/PhysRev.95.585.2.
- Audi, G.; Kondev, F. G.; Wang, M.; Huang, W. J.; Naimi, S. (2017). "The NUBASE2016 evaluation of nuclear properties" (PDF). Chinese Physics C. 41 (3): 030001. Bibcode:2017ChPhC..41c0001A. doi:10.1088/1674-1137/41/3/030001.
- Audi, Georges; Bersillon, Olivier; Blachot, Jean; Wapstra, Aaldert Hendrik (2003), "The NUBASE evaluation of nuclear and decay properties", Nuclear Physics A, 729: 3–128, Bibcode:2003NuPhA.729....3A, doi:10.1016/j.nuclphysa.2003.11.001
- Greenwood, Norman N.; Earnshaw, Alan (1984). Chemistry of the Elements. Oxford: Pergamon Press. p. 1262. ISBN 978-0-08-022057-4.
- Sonzogni, Alejandro. "Interactive Chart of Nuclides". National Nuclear Data Center: Brookhaven National Laboratory. Retrieved 6 June 2008.
- Zagrebaev, Valeriy; Karpov, Alexander; Greiner, Walter (2013). "Future of superheavy element research: Which nuclei could be synthesized within the next few years?" (PDF). Journal of Physics. 420 (1): 012001. arXiv:1207.5700. Bibcode:2013JPhCS.420a2001Z. doi:10.1088/1742-6596/420/1/012001. S2CID 55434734.
- Luig, Heribert; Keller, Cornelius; Wolf, Walter; Shani, Jashovam; Miska, Horst; Zyball, Alfred; Gervé, Andreas; Balaban, Alexandru T.; Kellerer, Albrecht M.; Griebel, Jürgen (2000). "Radionuclides". Ullmann's Encyclopedia of Industrial Chemistry. doi:10.1002/14356007.a22_499. ISBN 978-3527306732.
- "High Flux Isotope Reactor". Oak Ridge National Laboratory. Retrieved 23 September 2010.
- Porter, C. E.; Riley, F. D., Jr.; Vandergrift, R. D.; Felker, L. K. (1997). "Fermium Purification Using Teva Resin Extraction Chromatography". Sep. Sci. Technol. 32 (1–4): 83–92. doi:10.1080/01496399708003188.
- Sewtz, M.; Backe, H.; Dretzke, A.; Kube, G.; Lauth, W.; Schwamb, P.; Eberhardt, K.; Grüning, C.; Thörle, P.; Trautmann, N.; Kunz, P.; Lassen, J.; Passler, G.; Dong, C.; Fritzsche, S.; Haire, R. (2003). "First Observation of Atomic Levels for the Element Fermium (Z=100)". Phys. Rev. Lett. 90 (16): 163002. Bibcode:2003PhRvL..90p3002S. doi:10.1103/PhysRevLett.90.163002. PMID 12731975. S2CID 16234935.
- Hoff, R. W.; Hulet, E. K. (1970). "Engineering with Nuclear Explosives". 2: 1283–1294. Cite journal requires
|journal=(help) - Choppin, G. R.; Harvey, B. G.; Thompson, S. G. (1956). "A new eluant for the separation of the actinide elements". J. Inorg. Nucl. Chem. 2 (1): 66–68. doi:10.1016/0022-1902(56)80105-X.
- Mikheev, N. B.; Kamenskaya, A. N.; Konovalova, N. A.; Rumer, I. A.; Kulyukhin, S. A. (1983). "High-speed method for the separation of fermium from actinides and lanthanides". Radiokhimiya. 25 (2): 158–161.
- Seaborg, p. 39
- Seaborg, p. 40
- United States Nuclear Tests July 1945 through September 1992 Archived June 15, 2010, at the Wayback Machine, DOE/NV--209-REV 15, December 2000
- Seaborg, p. 43
- Seaborg, p. 44
- Seaborg, p. 47
- Emsley, John (2011). Nature's Building Blocks: An A-Z Guide to the Elements (New ed.). New York, NY: Oxford University Press. ISBN 978-0-19-960563-7.
- Lundqvist, Robert; Hulet, E. K.; Baisden, T. A.; Näsäkkälä, Elina; Wahlberg, Olof (1981). "Electromigration Method in Tracer Studies of Complex Chemistry. II. Hydrated Radii and Hydration Numbers of Trivalent Actinides". Acta Chemica Scandinavica A. 35: 653–661. doi:10.3891/acta.chem.scand.35a-0653.
- Hussonnois, H.; Hubert, S.; Aubin, L.; Guillaumont, R.; Boussieres, G. (1972). Radiochem. Radioanal. Lett. 10: 231–238. Missing or empty
|title=(help) - Thompson, S. G.; Harvey, B. G.; Choppin, G. R.; Seaborg, G. T. (1954). "Chemical Properties of Elements 99 and 100". J. Am. Chem. Soc. 76 (24): 6229–6236. doi:10.1021/ja01653a004.
- Malý, Jaromír (1967). "The amalgamation behaviour of heavy elements 1. Observation of anomalous preference in formation of amalgams of californium, einsteinium, and fermium". Inorg. Nucl. Chem. Lett. 3 (9): 373–381. doi:10.1016/0020-1650(67)80046-1.
- Mikheev, N. B.; Spitsyn, V. I.; Kamenskaya, A. N.; Gvozdec, B. A.; Druin, V. A.; Rumer, I. A.; Dyachkova, R. A.; Rozenkevitch, N. A.; Auerman, L. N. (1972). "Reduction of fermium to divalent state in chloride aqueous ethanolic solutions". Inorg. Nucl. Chem. Lett. 8 (11): 929–936. doi:10.1016/0020-1650(72)80202-2.
- Hulet, E. K.; Lougheed, R. W.; Baisden, P. A.; Landrum, J. H.; Wild, J. F.; Lundqvist, R. F. (1979). "Non-observance of monovalent Md". J. Inorg. Nucl. Chem. 41 (12): 1743–1747. doi:10.1016/0022-1902(79)80116-5.
- Dictionary of Inorganic Compounds. 3 (1 ed.). Chapman & Hall. 1992. p. 2873. ISBN 0412301202.
- Mikheev, N. B.; Spitsyn, V. I.; Kamenskaya, A. N.; Konovalova, N. A.; Rumer, I. A.; Auerman, L. N.; Podorozhnyi, A. M. (1977). "Determination of oxidation potential of the pair Fm2+/Fm3+". Inorg. Nucl. Chem. Lett. 13 (12): 651–656. doi:10.1016/0020-1650(77)80074-3.
- Nugent, L. J. (1975). MTP Int. Rev. Sci.: Inorg. Chem., Ser. One. 7: 195–219. Missing or empty
|title=(help) - Samhoun, K.; David, F.; Hahn, R. L.; O'Kelley, G. D.; Tarrant, J. R.; Hobart, D. E. (1979). "Electrochemical study of mendelevium in aqueous solution: No evidence for monovalent ions". J. Inorg. Nucl. Chem. 41 (12): 1749–1754. doi:10.1016/0022-1902(79)80117-7.
- Koch, Lothar (2000). "Transuranium Elements". Transuranium Elements, in Ullmann's Encyclopedia of Industrial Chemistry. Wiley. doi:10.1002/14356007.a27_167. ISBN 978-3527306732.
Further reading
- Robert J. Silva: Fermium, Mendelevium, Nobelium, and Lawrencium, in: Lester R. Morss, Norman M. Edelstein, Jean Fuger (Hrsg.): The Chemistry of the Actinide and Transactinide Elements, Springer, Dordrecht 2006; ISBN 1-4020-3555-1, p. 1621–1651; doi:10.1007/1-4020-3598-5_13.
- Seaborg, Glenn T. (ed.) (1978) Proceedings of the Symposium Commemorating the 25th Anniversary of Elements 99 and 100, 23 January 1978, Report LBL-7701
- Gmelins Handbuch der anorganischen Chemie, System Nr. 71, Transurane: Teil A 1 II, p. 19–20; Teil A 2, p. 47; Teil B 1, p. 84.
External links
| Wikimedia Commons has media related to Fermium. |
| Look up fermium in Wiktionary, the free dictionary. |
- Fermium at The Periodic Table of Videos (University of Nottingham)