c-Met inhibitor
c-Met inhibitors are a class of small molecules that inhibit the enzymatic activity of the c-Met tyrosine kinase, the receptor of hepatocyte growth factor/scatter factor (HGF/SF). These inhibitors may have therapeutic application in the treatment of various types of cancers.[1]
Many c-Met inhibitors are currently in clinical trials. Crizotinib[2] and cabozantinib were the first to be approved by the U.S. FDA. Crizotinib received accelerated approval in 2011 for the treatment of patients with locally advanced or metastatic non-small cell lung cancer, while cabozantinib was approved in 2012 for the treatment of medullary thyroid cancer[3] and it has also started clinical trials for the treatment of several other types of cancer.
c-Met stimulates cell scattering, invasion, protection from apoptosis and angiogenesis.[4] c-Met is a receptor tyrosine kinase,[5] which can cause a wide variety of different cancers, such as renal, gastric and small cell lung carcinomas, central nervous system tumours, as well as several sarcomas [6] when its activity is dysregulated. Targeting the ATP binding site of c-Met by small molecules inhibitors is one strategy for inhibition of the tyrosine kinase.[7]
History
Early in the 1980s MET was described as the protein product of a transforming oncogene.[9] [10]
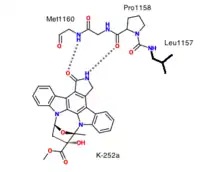
Initial attempts to identify ATP-competitive c-Met inhibitors in 2002 led to the discovery of K252a, a staurosporine-like inhibitor which blocks c-Met.[10][11] K252a was the first structure to be solved in complex with the unphosphorylated MET kinase domain. It forms two hydrogen bonds between the hinge and pyrralocarbazole subunit.[8]
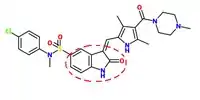
Later, series of more selective c-Met inhibitors were designed, where an indolin-2-one core (encircled in figure 1) was present in several kinase inhibitors. SU-11274 was evolved by substitution at the 5-position of the indolinone [9] and by adding a 3,5-dimethyl pyrrole group, PHA-665752 was evolved [11] – a second-generation inhibitor with better potency and activity.[10]
Interest in this field has risen rapidly since 2007 and over 70 patent applications had been published in mid-2009.[10]
Intensive efforts have been exerted in the pharmaceutical industry following the acceptance of c-Met as a suitable target for cancer therapy. 20 crystal structures with and without ligands have been published and in 2010 nearly a dozen small molecule c-Met inhibitors have been tested clinically.[12]
Introduction
Receptor tyrosine kinases (RTKs) are a vital element in regulating many intracellular signal transduction pathways.[13] Met tyrosine kinase is the receptor for hepatocyte growth factor (HGF), also known as scatter factor (SF). HGF is mostly expressed on epithelial cells and mesenchymal cells, for example smooth muscle cells and fibroblasts).[10][11] HGF is normally active in wound healing, liver regeneration, embryo and normal mammalian development,[10] organ morphogenesis.[11]
c-Met dysregulation can be due to overexpression, gene amplification, mutation, a ligand-dependent auto- or paracrine loop or an untimely activation of RTK.[10][13] All these factors affect the survival of cells, their proliferation and motility. They also lead to cancers and resistance to therapies which aim to treat them.[13] Patients with aberrant c-Met activity usually have a poor prognosis, aggressive disease, increased metastasis and shortened survival.[10] This is why targeting the HGF/c-MET signalling pathway has been untaken as a treatment for cancer,[10][13] and several different therapeutic approaches are being clinically tested. A variety of approaches have been used to target c-Met, each focusing on one of the serial steps that regulate c-Met activation by antibodies, peptide agonists,[4][10] decoy receptors and other biologic inhibitors[14] or small molecules inhibitors.[10]
Structure and function
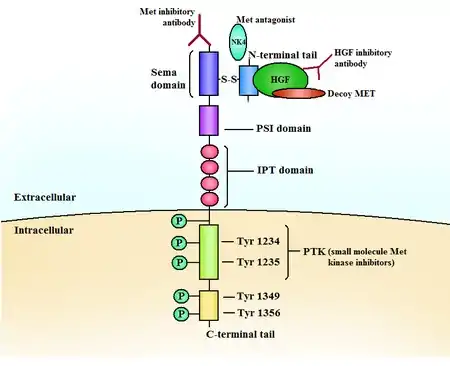
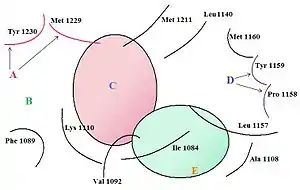
The c-Met RTK subfamily is different in structure to many other RTK families: The mature form has an extracellular α-chain (50kDa) and a transmembrane β-chain (140kDa) that are linked together by a disulfide bond. The beta chain contains the intracellular tyrosine kinase domain and a tail on the C-terminal which is vital for the docking of substrates and downstream signalling.[10] [17]
HGF is the natural high-affinity ligand for Met.[10][11][17] Its N-terminal region binds to Met and receptor dimerization as well as autophosphorylation of two tyrosines occur in the activation loop (A-loop) in the kinase domain of Met.[10]
Phosphorylation occurs in tyrosines close to the C-terminus, creating a multi-functional docking site[10][18] which recruits adaptor proteins and leads to downstream signalling. The signaling is mediated by Ras/Mapk, PI3K/Akt, c-Src and STAT3/5 and include cell proliferation, reduced apoptosis, altered cytoskeletal function and more.
The kinase domain usually consists of a bi-lobed structure, where the lobes are connected with a hinge region, adjacent to the very conserved ATP binding site.[10]
Development
Using information from the co-crystal structure of PHA-66752 and c-Met, the selective inhibitor PF-2341066 was designed. It was undergoing Phase I/II clinical trials in 2010. Changing a series of 4-phenoxyquinoline compounds with an acyl thiourea group led to compounds with c-Met activity, e.g. quinoline.[10] This was a key step in the progress of c-Met inhibitor development in that the acyl binding gives the terminal aryl group the ability to penetrate a deep hydrophobic pocket and so it enhances the potency of the compounds. Alternatives to the acyl thiourea linkage have been found, which have a pyrimidone group, as in AM7.[19]
AM7 and SU11274 offered the first proof that relatively selective c-Met inhibitors could be identified and that the inhibition leads to an anti-tumour effect in vivo. When the co-crystal structures of AM7 and SU11274 with c-Met were compared, they were found to be different: SU-11274 binds adjacent to the hinge region with a U-shaped conformation; but AM7 binds to c-Met in an extended conformation which spans the area from the hinge region to the C-helix. It then binds in a hydrophobic pocket. c-Met assumes an inactive, unphosphorylated conformation with AM7, which can bind to both phosphorylated and unphosphorylated conformations of the kinase.[20]
Due to these two different types of binding, small molecule Met inhibitors have been divided into two classes; class I (SU-11274-like) and class II (AM7-like).[20] There is however another type of small-molecule inhibitors, which does not fit into either of the two classes; a non-competitive ATP inhibitor that binds in a different way to the other two.[21]
The small molecule inhibitors vary in selectivity, are either very specific or have a broad selectivity. They are either ATP competitive or non-competitive.[12]
ATP-competitive small molecule c-Met inhibitors
Even though the two classes are structurally different, they do share some properties: They both bind at the kinase hinge region (although they occupy different parts of the c-Met active site[20]) and they all aim to mimic the purine of ATP. BMS-777607 and PF-02341066 have a 2-amino-pyridine group, AMG-458 has a quinoline group and MK-2461 has a tricyclic aromatic group.[22]
Class I
Class I inhibitors have many different structures,[12] are relatively selective and have a U-shaped conformation[10] and binds to the activation loop of c-Met.[12]
Structure-activity relationship of Class I inhibitors
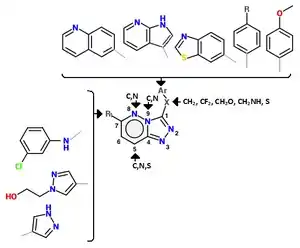
A series of triazolotriazines was discovered, which showed great promise as a c-MET inhibitors. Structure activity relationship (SAR) implies the necessity of an aryl group linked to the triazine ring and an appropriate hydrogen bond acceptor (e.g. hydroxyl group) attached to the pendant benzyl ring but it seems like the phenol acts as a hinge binder (with Met1160) and that the triazine interacts with Tyr1230.[12] A number of similar analogues were found and assayed. Structurally similar series of c-Met inhibitors in which a phenolic hinge binding element was linked to an arylamino-triazolopyridazine or aryl-triazolothiapyridazine. One-atom linker was more efficient than a two-atom linker and that substitution at the benzylic position seemed to be tolerated. Compounds with heterocyclic hinge binding elements (quinoline, pyridine, azaindole) linked to fused, nitrogen-dense heteroaromatics (triazolopyridazines, triazolopyrazines and triazolotriazines) have been described.[12] See figure 4 for details.[12]
Examples of Class I inhibitors
JNJ-38877605, which contains a difluoro methyl linker and a bioavailable quinoline group, was undergoing clinical trials of Phase I for advanced and refractory solid tumours in 2010.[12]
PF-04217903, an ATP-competitive and exceptionally selective compound, has an N-hydroxyethyl pyrazole group tethered to C-7 of the triazolopyrazine. It was undergoing phase I clinical trials in 2010.[12]
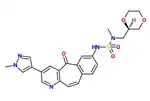
The SAR of the unique kinase inhibitor scaffold with powerful c-Met inhibitory activity, MK-2461, has been explored.[23] The pyridine nitrogen is necessary for inhibition activity and central ring saturation reduced potency.[12] Planarity of the molecule has proven to be essential for maximum potency.[23] Cyclic ethers balance acceptable cell-based activities and pharmacokinetic characteristics. The following elements are thought to be key in the optimization process:
1) Aryl groups at the 7-position, as if to maximize hydrophobic packing and planarity,
2) The tight SAR upon the addition of a sulfonamide group and
3) The relatively flat SAR of solvent-exposed groups.
Often, oncogenic mutations of c-Met cause a resistance to small molecule inhibitors. An MK-2461 analog was therefore tested against a variety of c-Met mutants but proved to be no less potent against them. This gives the molecule a big advantage as a treatment for tumours caused by c-Met dysregulation.[23] MK-2461 was undergoing phase I dose escalation trials in 2010.[12]
Class II
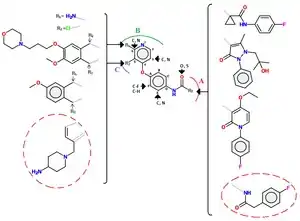
Class II inhibitors are usually not as selective as those of class I.[10] Urea groups are also a common feature of class II inhibitors, either in cyclic or acyclic forms. Class II of inhibitors contains a number of different molecules, a common scaffold of which can be seen in figure 4.[12]
Structure-activity relationship of Class II inhibitors
Series of quinoline c-Met inhibitors with an acylthiourea linkage have been explored. Multiple series of analogs have been found with alternative hinge binding groups (e.g. replacement of the quinoline group), replacement of the thiourea linkage (e.g. malonamide, oxalamide, pyrazolones) and constraining of the acyclic acylthiourea structure fragment with various aromatic heterocycles. Further refinement included the blocking of the p-position of the pendant phenyl ring with a fluorine atom.[12] Example of interactions between c-Met and a small molecules (marked in a red circle) of class II are as follows: The scaffold of c-Met lodges into the ATP pocket by three key hydrogen bonds, the terminal amine interacts with the ribose pocket (of ATP), the terminal 4-fluorophenyl group is oriented in a hydrophobic pocket and pyrrolotriazine plays the role of the hinge-binding group.[12]
Examples of Class II inhibitors
In phase II clinical trials, GSK 1363089 (XL880, foretinib) was well tolerated. It led to slight regressions or stable disease in patients with papillary renal carcinoma and poorly differentiated gastric cancer.[12]
AMG 458 is a potent small molecule c-MET inhibitor which proved to have more than a 100-fold selectivity for c-MET across a panel of 55 kinases. Also, AMG 458 was 100% bioavailable across species and the intrinsic half-life increased with higher mammals.[12]
ATP non-competitive small molecule c-Met inhibitors
Tivantinib
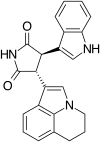
Tivantinib (ARQ197) is a selective, orally bioavailable,[17][21] clinically advanced low-molecular weight and well-tolerated c-MET inhibitor, which is currently in Phase III clinical trials in non-small cell lung cancer patients.[21] ARQ197 is a non-ATP competitive c-MET autophosphorylation inhibitor with a high selectivity for the unphosphorylated conformation of the kinase.[17][21] Tivantinib cuts off the interactions between the key catalytic residues.[21] The structure of tivantinib in complex with the c-Met kinase domain shows that the inhibitor binds a conformation that is distinct from published kinase structures. Tivantinib strongly inhibits c-Met autoactivation by selectively targeting the inactive form of the kinase between the N- and C- lobes and occupies the ATP binding site.[21]
Clinical trials and regulatory approvals
Status as of 2010
Since the discovery of Met and HGF, much research interest has focused on their roles in cancer. The Met pathway is one of the most frequently dysregulated pathways in human cancer.[17] Increased understanding of the binding modes and structural design brings us closer to the use of other protein interactions and binding pockets, creating inhibitors with alternative structures and optimized profiles.[10]
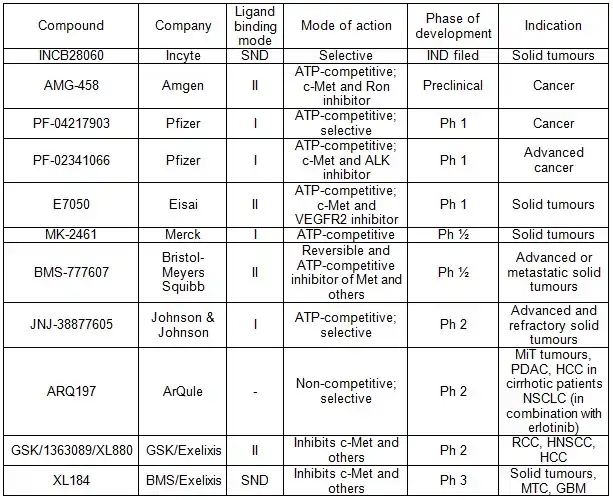
As of 2010 over a dozen Met pathway inhibitors, with varying kinase selectivity profiles ranging from highly selective to multi-targeted,[12] have been studied in the clinic and good progress has been achieved [17] (See table 1). (e.g. XL184(Cabozantinib), XL880, ARQ197 )
The use of c-Met inhibitors with other therapeutic agents could be crucial for overcoming potential resistance as well as for improving overall clinical benefit. Met pathway inhibitors might be used in combination with other treatments, including chemo-, radio- or immunotherapy as well as different Met pathway inhibitor, f.ex. in with HGF and Met biological antagonists or antibodies against HGF and MET.[17] Still, the risk of accumulated toxicity and interactions with other drugs remains.[10]
Since 2010
In 2011 PF-02341066 (now named crizotinib) was approved by US FDA for some non-small cell lung cancers.
In 2012 XL184/cabozantinib gained FDA approval to treat medullary thyroid cancer, and in 2016 it gained FDA and EU approval to treat kidney cancer.
Research on other inhibitors
Tepotinib, (MSC 2156119J),[24]
has reported phase II clinical trial results on lung cancer.[25] Tepotinib was granted breakthrough therapy designation by the U.S. Food and Drug Administration (FDA) in September 2019.[26] It was granted orphan drug designation in Japan in November 2019, and in Australia in September 2020.[27]
See also
External links
References
- Liu X, Newton RC, Scherle PA (September 2011). "Development of c-MET pathway inhibitors". Expert Opin Investig Drugs. 20 (9): 1225–41. doi:10.1517/13543784.2011.600687. PMID 21740293. S2CID 24415851.
- Kazandjian, D; et al. (Oct 2014). "FDA approval summary: crizotinib for the treatment of metastatic non-small cell lung cancer with anaplastic lymphoma kinase rearrangements". Oncologist. 19 (10): e5-11. doi:10.1634/theoncologist.2014-0241. PMC 4201002. PMID 25170012.
- "FDA approves Cometriq to treat rare type of thyroid cancer". 29 November 2012.
- Comoglio PM, Giordano S, Trusolino L (June 2008). "Drug development of MET inhibitors: targeting oncogene addiction and expedience". Nature Reviews Drug Discovery. 7 (6): 504–16. doi:10.1038/nrd2530. PMID 18511928. S2CID 24601127.
- Maulik G, Shrikhande A, Kijima T, Ma PC, Morrison PT, Salgia R (February 2002). "Role of the hepatocyte growth factor receptor, c-Met, in oncogenesis and potential for therapeutic inhibition". Cytokine Growth Factor Rev. 13 (1): 41–59. doi:10.1016/S1359-6101(01)00029-6. PMID 11750879.
- Davis IJ, McFadden AW, Zhang Y, Coxon A, Burgess TL, Wagner AJ, Fisher DE (January 2010). "Identification of the receptor tyrosine kinase c-Met and its ligand, hepatocyte growth factor, as therapeutic targets in clear cell sarcoma". Cancer Res. 70 (2): 639–45. doi:10.1158/0008-5472.CAN-09-1121. PMC 2807989. PMID 20068147.
- Porter J, Lumb S, Franklin RJ, Gascon-Simorte JM, Calmiano M, Riche KL, Lallemand B, Keyaerts J, Edwards H, Maloney A, Delgado J, King L, Foley A, Lecomte F, Reuberson J, Meier C, Batchelor M (May 2009). "Discovery of 4-azaindoles as novel inhibitors of c-Met kinase". Bioorg. Med. Chem. Lett. 19 (10): 2780–4. doi:10.1016/j.bmcl.2009.03.110. PMID 19369077.
- Schiering N, Knapp S, Marconi M, Flocco MM, Cui J, Perego R, Rusconi L, Cristiani C (October 2003), "Crystal structure of the tyrosine kinase domain of the hepatocyte growth factor receptor c-Met and its complex with the microbial alkaloid K-252a", Proc. Natl. Acad. Sci. U.S.A., 100 (22): 12654–12659, Bibcode:2003PNAS..10012654S, doi:10.1073/pnas.1734128100, PMC 240673, PMID 14559966
- Sattler M, Pride YB, Ma P, Gramlich JL, Chu SC, Quinnan LA, Shirazian S, Liang CX, Podar K, Christensen JG, Salgia R (September 2003), "A novel small molecule Met inhibitor induces apoptosis in cells transformed by the oncogenic TPR-MET tyrosine kinase", Cancer Research, 63 (17): 5462–5469, PMID 14500382
- Porter, J (February 2010), "Small molecule c-Met kinase inhibitors: a review of recent patents", Expert Opinion on Therapeutic Patents, 20 (2): 159–177, doi:10.1517/13543770903514137, PMID 20100000, S2CID 22743228
- Christensen JG, Schreck R, Burrows J, Kuruganti P, Chan E, Le P, Chen J, Wang XY, Ruslim L, Blake R, Lipson KE, Ramphal J, Do S, Cui JR, Cherrington JM, Mendel DB (November 2003), "A selective small molecule inhibitor of c-Met kinase inhibits c-Met dependent phenotypes in vitro and exhibits cytoreductive antitumor activity in vivo", Cancer Research, 63 (21): 7345–55, PMID 14612533
- Underiner TL, Herbertz T, Miknyoczki SJ (January 2010), "Discovery of Small Molecule c-Met Inhibitors: Evolution and Profiles of Clinical Candidates", Anti-Cancer Agents in Medicinal Chemistry, 10 (1): 7–27, doi:10.2174/1871520611009010007, PMID 20015007
- Sattler M, Salgia R (April 2009), "The Met axis as a therapeutic target", Update on Cancer Therapeutics, 3 (3): 109–118, doi:10.1016/j.uct.2009.01.001, PMC 2847295, PMID 20368753
- Christensen JG; Burrows J; Salgia R. (July 2005), "c-Met as a target for human cancer and characterization of inhibitors for therapeutic intervention", Cancer Letters, 225 (1): 1–26, doi:10.1016/j.canlet.2004.09.044, PMID 15922853
- Knudsen BS, Woude GV (February 2008), "Showering c-MET-dependent cancers with drugs", Current Opinion in Genetics & Development, 18 (1): 87–96, doi:10.1016/j.gde.2008.02.001, PMID 18406132
- Donald P. Bottaro; Megan Peach; Marec Nicklaus; Terrence Burke, JR.; Gagani Athauda; Sarah Choyke; Alessio Guibellino; Nelly Tan; Zhen-Dan Shi (August 2011), "Compositions and methods for inhibition of hepatocyte growth factor receptor c-Met signaling", United States Patent Application Publication
- Liu XD, Newton RC, Scherle PA (January 2010), "Developing c-MET pathway inhibitors for cancer therapy: progress and challenges", Trends in Molecular Medicine, 16 (1): 37–45, doi:10.1016/j.molmed.2009.11.005, PMID 20031486
- Kung PP, Funk L, Meng J, Alton G, Padrique E, Mroczkowski B (June 2008), "Structure activity relationships of quinoline-containing c-Met inhibitors", European Journal of Medicinal Chemistry, 43 (8): 1321–1329, doi:10.1016/j.ejmech.2007.08.011, PMID 17964000
- Bellon SF; Kaplan-Lefko P; Yang YJ; Zhang YH; Moriguchi J; Rex K; Johnson CW; Rose PE; Long AM; O‘Connor AB; Gu Y; Coxon A; Kim TS; Tasker A; Burgess TL; Dussault I (February 2008), "c-Met inhibitors with novel binding mode show activity against several hereditary papillary renal cell carcinoma-related mutations", Journal of Biological Chemistry, 283 (5): 2675–2683, doi:10.1074/jbc.M705774200, PMID 18055465
- Dussault I, Bellon SF (February 2009), "From concept to reality: the long road to c-Met and RON receptor tyrosine kinase inhibitors for the treatment of cancer", Anti-Cancer Agents in Medicinal Chemistry, 9 (2): 221–229, doi:10.2174/187152009787313792, PMID 19199866
- Eathiraj S, Palma R, Volckova E, Hirschi M, France DS, Ashwell MA, Chan TC (June 2011), "Discovery of a Novel Mode of Protein Kinase Inhibition Characterized by the Mechanism of Inhibition of Human Mesenchymal-epithelial Transition Factor (c-Met) Protein Autophosphorylation by ARQ 197", Journal of Biological Chemistry, 286 (23): 20666–20676, doi:10.1074/jbc.M110.213801, PMC 3121448, PMID 21454604
- Allen JV, Bardelle C, Blades K, Buttar D, Chapman L, Colclough N, Dossetter AG, Garner AP, Girdwood A, Lambert C, Leash AG, Law B, Major J, Plant H, Slater AM (September 2011), "The discovery of benzanilides as c-Met receptor tyrosine kinase inhibitors by a directed screening approach", Bioorganic & Medicinal Chemistry Letters, 21 (18): 5224–5229, doi:10.1016/j.bmcl.2011.07.047, PMID 21835616
- Katz JD, Jewell JP, Guerin DJ, Lim J, Dinsmore CJ, Deshmukh SV, Pan BS, Marshall CG, Lu W, Altman MD, Dahlberg WK, Davis L, Falcone D, Gabarda AE, Hang GZ, Hatch H, Holmes R, Kunii K, Lumb KJ, Lutterbach B, Mathvink R, Nazef N, Patel SB, Qu XL, Reilly JF, Rickert KW, Rosenstein C, Soisson SM, Spencer KB, Szewczak AA, Walker D, Wang WX, Young J, Zeng QW (June 2011), "Discovery of a 5H-Benzo[4,5]cyclohepta[1,2-b]pyridin-5-one (MK-2461) Inhibitor of c-Met Kinase for the Treatment of Cancer", Journal of Medicinal Chemistry, 54 (12): 4092–4108, doi:10.1021/jm200112k, PMID 21608528
- Tepotinib With Gefitinib in Subjects With Locally Advanced or Metastatic Non-small Cell Lung Cancer (NSCLC) (INSIGHT)
- Phase II trial of the c-Met inhibitor tepotinib in advanced lung adenocarcinoma with MET exon 14 skipping mutations. 2017
- "Tepotinib Breakthrough Therapy". Merck KGaA, Darmstadt, Germany (Press release). 11 September 2019. Retrieved 8 November 2020.
- "Orphan Drug Designation". Merck KGaA, Darmstadt, Germany (Press release). 20 November 2019. Retrieved 8 November 2020.