WNK4
Serine/threonine protein kinase WNK4 also known as WNK lysine deficient protein kinase 4 or WNK4, is an enzyme that in humans is encoded by the WNK4 gene.[5] Missense mutations cause a genetic form of pseudohypoaldosteronism type 2, also called Gordon syndrome.


WNK4 is a member of a serine/threonine kinase family that comprises four members. The family is so named because unlike other serine/threonine kinases, WNKs are characterized by the lack of the lysine in the subdomain II of the catalytic domain.[6] Instead, a lysine in the β2 strand of subdomain I of the catalytic domain is responsible for the kinase activity.[6]
The WNK4 gene is located on chromosome 17q21-q22. It produces a 1,243-amino acid protein encoded by a 3,732-nucleotide open reading frame within a 4 kb cDNA transcript.[7] WNK4 protein is highly expressed in the distal convoluted tubule (DCT) and the cortical collecting duct (CDD) of the kidney.[7] WNK4 is also present in the brain, lungs, liver, heart, and colon of various mammalian species.[8][9]
Gene mutations in WNK4 has been found in patients with pseudohypoaldosteronism type II (PHAII),[7] also known as familial hyperkalemic hypertension (FHHt)[10] or Gordon syndrome.[11] PHAII is an autosomal dominant hereditary disease characterized by hyperkalemia, hypertension, and metabolic acidosis. WNK4 plays a critical role in the regulation of various transporters and channels in the kidney. PHAII-causing mutations in WNK4 result in the dysregulation of renal sodium and potassium transporters and channels, leading to defects in sodium and potassium retention by the kidney, and in turn, elevated blood pressure and potassium level (hyperkalemia).
Structure
The tertiary structure of WNK4 has not been elucidated to date. Nevertheless, several individual domain structures of the protein are identified. These include a kinase domain near the amino terminus followed by an autoinhibitory domain, an acidic motif, two coiled-coil domains, and a calmodulin-binding domain in the C-terminal segment (Fig. 1). The structures for the kinase and autoinhibitory domains of WNK1 have been revealed. The high level of structural similarity between WNK4 and WNK1 allows us to deduce key structural details of WNK4 based on the insights gained from the corresponding regions within WNK1. The kinase domain of WNK4 has an 83% sequence identity with that of WNK1. The overall fold of the kinase domain of WNK1 resembles those of other protein kinases that have a typical dual-domain architecture.[12] The C-terminal domain of WNK4 bears a high degree of similarity to other kinases within the family. On the other hand, the N-terminal domain is unique in having a six-stranded instead of a five-stranded β sheet to form a complete β barrel.[12]
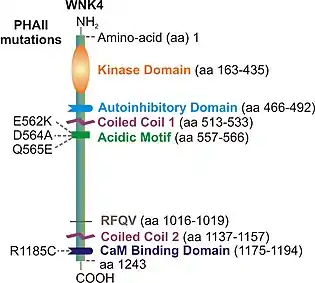
A chloride ion binding site has been identified in the region 320DLG323 of the kinase domain in WNK4.[13] The binding of chloride Cl− in this region inhibits the activation of WNK4. The autoinhibitory domain is a homolog of the RFXV-binding PASK/FRAY homology 2 (PF2) domain.[14] Structural studies have revealed that the autoinhibitory domain consists of three β-strands and two α-helices.[15] Notably, the RFXV‐binding groove is formed by the β3-αA interface of WNK proteins where RFXV peptide ligand interacts directly with residues Phe524, Asp531, and Glu539 of WNK1.[15] The interaction between the RFXV motif and the autoinhibitory domain makes it possible for the C-terminal region of WNK4 to be in close proximity of the kinases domain and subsequently regulate its activity.
Function
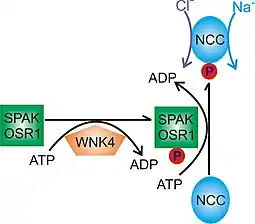
As a typical kinase, WNK4 accomplishes the phosphorylation of its substrate proteins by adding a phosphate moiety in an ATP-dependent manner. This structural modification usually results in functional alterations of downstream substrates. Some currently known substrates of WNK4 includes kinases STE20-serine-proline alanine-rich kinase (SPAK) and oxidative stress response 1 kinase (OSR1), which in turn can phosphorylate and activate the thiazide-sensitive sodium-chloride cotransporter (NCC)[16][17] (Fig. 2). Similarly, WNK4 activates NKCC1 and deactivate KCC2 through a SPAK-dependent mechanism.[18] The kinase activity of WNK4 has been demonstrated in vitro using the WNK4 kinase domain purified from E. coli.[19] This phosphorylation cascade is critical in regulating sodium and potassium homeostasis dysregulation which is tied to the pathogenesis of PHAII.
In addition to NCC, WNK4 also regulates multiple ions channels and cotransporters in the kidney through various mechanisms. These include epithelial Na+ channel (ENaC), renal outer medullary potassium channel (ROMK), transient receptor potential vanilloid member 4 and 5 (TRPV4/5, calcium channels), Na-K-2Cl cotransporter 1 and 2 (NKCC1/2), K+-Cl− cotransporter type 2 (KCC2), and other channels/transporters. WNK4 inhibits the functions of ENaC, ROMK, and TRPV4 by reducing the total and cell surface expression of these channels.[20][21][22] WNK4 enhances TRPV5 by increasing its forward trafficking to the plasma membrane in a kinase-dependent manner.[23] The inhibitory effect of WNK4 on ROMK is reversed by serum and glucocorticoid kinase 1 (SGK1) or by a corresponding phosphomimetic S1169D mutation on WNK4.[21] The N-terminal segment of WNK4 containing the kinase domain and acidic motif is required for the WNK4-mediated inhibition of ROMK.[24] The second coiled-coil domain of WNK4 mediates the downregulation of TRPV4. WNK4 and calcium-binding protein 39 (Cab39) act together to activate transporters NKCC1 and NKCC2.[25]
Role in pseudohypoaldosteronism type 2
Dysregulation of WNK4 kinase activity
In 2001, four missense mutations in the WNK4 gene were identified in patients with pseudohypoaldosteronism type 2 (PHAII) (Fig. 1).[7] Three of these mutations (E562K, D564A, and Q565E) are charge-changing substitutions in the acidic motif of WNK4, which are conserved among all members of the WNK family in human and rodent species. The fourth substitution (R1185C) is located in the calmodulin-binding domain near the second coiled-coil domain. Few other PHAII mutations in WNK4 have also been reported. Examples of these mutations include E560G,[26] P561L,[27] and D564H,[28] all of which are located close to or in the acidic motif; and K1169E [29] which is located between the coiled-coil 2 and the calmodulin-binding domain.
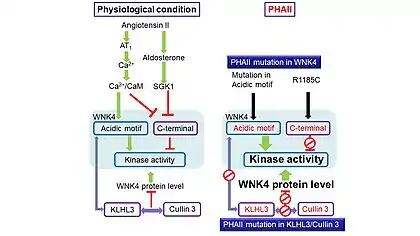
The PHAII mutations appear to disrupt the mechanisms underlying Ca2+-sensitivity of WNK4 kinase. Two mechanisms are important in this regard. First, the PHAII-causing mutations in the acidic motif make the kinase domain insensitive to Ca2+ concentration. The acidic motif of WNK4 potentially acts as a Ca2+ sensor, and WNK4 kinase activity rises when Ca2+ concentration is elevated. This has been demonstrated using isolated WNK4 kinase domain truncated to contain the acidic motif.[19] The kinase activity is elevated when a PHAII-causing mutation is present in the acidic motif, similar to what is observed in a Ca2+-binding state (Fig. 3). Second, the WNK4 C-terminal region containing the calmodulin-binding domain and multiple SGK1 phosphorylation sites inhibits the WNK4 activity at the resting state.[30] However, when Ca2+ levels are elevated, Ca2+/calmodulin complex binds to the C-terminal region, derepressing WNK4 kinase activity. Additionally, the RFXV motif is believed to interact with the autoinhibitory domain and subsequently triggers a conformational change that brings the C-terminal and kinase domain close for the inhibitory effect to take place. Angiotensin II increases the SPAK phosphorylation and activates NCC through a WNK-dependent mechanism.[31] The activation of SPAK and NCC by angiotensin II is abrogated by WNK4 knockdown.[32] Activation of angiotensin II receptor AT1 couples to Gq/11 to activate phospholipase C and to increase the intracellular Ca2+ concentration. An increase in Ca2+ concentration then elevates WNK4 activity through mechanisms described above (Fig. 3, left panel). The PHAII-causing mutations in the acidic motif and the R1185C mutation in the calmodulin-binding domain constitutively activate the WNK4 kinase domain allowing it to function despite the absence of angiotensin II (Fig. 3, right panel).
Angiotensin II stimulates the secretion of aldosterone, which induces SGK1. SGK1 influences both the WNK-SPAK-NCC [33] and SGK1-ENaC signaling cascades.[34] There are multiple SGK1 phosphorylation sites in the C-terminal region of WNK4 located within or close to the calmodulin-binding domain. SGK1-mediated phosphorylation of these sites is thought to disrupt the effect of the C-terminal inhibitory domain and concomitantly increase WNK4 kinase activity.[30] The alteration of SGK1 phosphorylation by the R1185C mutation is another indication that the mutation disrupts the C-terminal inhibitory mechanism in WNK4 (Fig. 3, right panel).
Dysregulation of WNK4 abundance
Besides WNK1 and WNK4, mutations in two other genes, CUL3 (encoding Cullin 3) and KLHL3 (encoding Kelch Like Family Member 3) have been found in patients with PHAII.[35][36] These two proteins are part of the ubiquitin E3 ligase complex involved in the ubiquitin-mediated degradation of WNK1 and WNK4. The PHAII-causing mutations in KLHL3 and cullin 3 prevent the interactions of these proteins with each other and with WNK1/4. The mutations in these proteins impair the degradation of WNK1/4. This in turn increases the protein abundance of WNK1/4 and concomitantly enhances the total kinase activity.[37] The increased WNK4 kinase activity leads to the hyperactivation of NCC through WNK4-SPAK and/or the OSR1-NCC cascades, ultimately resulting in the retention of sodium and potassium by the kidney.
Elevated WNK4 activity
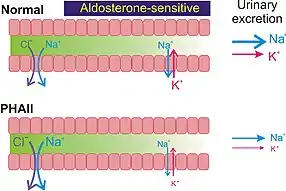
The primary effect of the elevated WNK4 kinase activity is the increase of NCC-mediated sodium reabsorption in the distal convoluted tubule of the kidney. The increase in sodium reabsorption in this segment of the nephron reduces the sodium load in the collecting duct, where sodium reabsorption by the ENaC provides the driving force for potassium secretion through ROMK (Fig. 4). The sodium reabsorption by hyperactive NCC overrides the loss of reabsorption by ENaC, and the net effect is moderate sodium retention. Over time, this potentially contributes to the elevated blood pressure observed in PHAII patients. The reduction of potassium secretion by ROMK contributes to the development of hyperkalemia. The direct effects of the elevated WNK4 activity on other channels and transporters, such as ENaC, ROMK, and Ca2+-activated maxi K+ channels, may also contribute to the pathogenesis of PHAII; however, the primary features of PHAII could be explained by the gain-of-function of NCC.
References
- GRCh38: Ensembl release 89: ENSG00000126562 - Ensembl, May 2017
- GRCm38: Ensembl release 89: ENSMUSG00000035112 - Ensembl, May 2017
- "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Entrez Gene: WNK4 WNK lysine deficient protein kinase 4".
- Xu B, English JM, Wilsbacher JL, Stippec S, Goldsmith EJ, Cobb MH (June 2000). "WNK1, a novel mammalian serine/threonine protein kinase lacking the catalytic lysine in subdomain II". The Journal of Biological Chemistry. 275 (22): 16795–801. doi:10.1074/jbc.275.22.16795. PMID 10828064.
- Wilson FH, Disse-Nicodème S, Choate KA, Ishikawa K, Nelson-Williams C, Desitter I, et al. (August 2001). "Human hypertension caused by mutations in WNK kinases". Science. 293 (5532): 1107–12. doi:10.1126/science.1062844. PMID 11498583. S2CID 22700809.
- Kahle KT, Gimenez I, Hassan H, Wilson FH, Wong RD, Forbush B, et al. (February 2004). "WNK4 regulates apical and basolateral Cl- flux in extrarenal epithelia". Proceedings of the National Academy of Sciences of the United States of America. 101 (7): 2064–9. doi:10.1073/pnas.0308434100. PMID 14769928.
- Veríssimo F, Jordan P (September 2001). "WNK kinases, a novel protein kinase subfamily in multi-cellular organisms". Oncogene. 20 (39): 5562–9. doi:10.1038/sj.onc.1204726. PMID 11571656.
- Arnold JE, Healy JK (September 1969). "Hyperkalemia, hypertension and systemic acidosis without renal failure associated with a tubular defect in potassium excretion". The American Journal of Medicine. 47 (3): 461–72. doi:10.1016/0002-9343(69)90230-7. PMID 5808659.
- Gordon RD, Geddes RA, Pawsey CG, O'Halloran MW (November 1970). "Hypertension and severe hyperkalaemia associated with suppression of renin and aldosterone and completely reversed by dietary sodium restriction". Australasian Annals of Medicine. 19 (4): 287–94. doi:10.1111/imj.1970.19.4.287. PMID 5490655.
- Min X, Lee BH, Cobb MH, Goldsmith EJ (July 2004). "Crystal structure of the kinase domain of WNK1, a kinase that causes a hereditary form of hypertension". Structure. 12 (7): 1303–11. doi:10.1016/j.str.2004.04.014. PMID 15242606.
- Piala AT, Moon TM, Akella R, He H, Cobb MH, Goldsmith EJ (May 2014). "Chloride sensing by WNK1 involves inhibition of autophosphorylation". Science Signaling. 7 (324): ra41. doi:10.1126/scisignal.2005050. PMC 4123527. PMID 24803536.
- Xu BE, Min X, Stippec S, Lee BH, Goldsmith EJ, Cobb MH (December 2002). "Regulation of WNK1 by an autoinhibitory domain and autophosphorylation". The Journal of Biological Chemistry. 277 (50): 48456–62. doi:10.1074/jbc.M207917200. PMID 12374799.
- Moon TM, Correa F, Kinch LN, Piala AT, Gardner KH, Goldsmith EJ (April 2013). "Solution structure of the WNK1 autoinhibitory domain, a WNK-specific PF2 domain". Journal of Molecular Biology. 425 (8): 1245–52. doi:10.1016/j.jmb.2013.01.031. PMID 23376100.
- Vitari AC, Deak M, Morrice NA, Alessi DR (October 2005). "The WNK1 and WNK4 protein kinases that are mutated in Gordon's hypertension syndrome phosphorylate and activate SPAK and OSR1 protein kinases". The Biochemical Journal. 391 (Pt 1): 17–24. doi:10.1042/BJ20051180. PMC 1237134. PMID 16083423.
- Moriguchi T, Urushiyama S, Hisamoto N, Iemura S, Uchida S, Natsume T, et al. (December 2005). "WNK1 regulates phosphorylation of cation-chloride-coupled cotransporters via the STE20-related kinases, SPAK and OSR1". The Journal of Biological Chemistry. 280 (52): 42685–93. doi:10.1074/jbc.M510042200. PMID 16263722.
- Gagnon KB, England R, Delpire E (January 2006). "Volume sensitivity of cation-Cl- cotransporters is modulated by the interaction of two kinases: Ste20-related proline-alanine-rich kinase and WNK4". American Journal of Physiology. Cell Physiology. 290 (1): C134-42. doi:10.1152/ajpcell.00037.2005. PMID 15930150.
- Na T, Wu G, Peng JB (March 2012). "Disease-causing mutations in the acidic motif of WNK4 impair the sensitivity of WNK4 kinase to calcium ions". Biochemical and Biophysical Research Communications. 419 (2): 293–8. doi:10.1016/j.bbrc.2012.02.013. PMC 3358818. PMID 22342722.
- Yu L, Cai H, Yue Q, Alli AA, Wang D, Al-Khalili O, et al. (July 2013). "WNK4 inhibition of ENaC is independent of Nedd4-2-mediated ENaC ubiquitination". American Journal of Physiology. Renal Physiology. 305 (1): F31-41. doi:10.1152/ajprenal.00652.2012. PMC 3725674. PMID 23594824.
- Ring AM, Leng Q, Rinehart J, Wilson FH, Kahle KT, Hebert SC, Lifton RP (March 2007). "An SGK1 site in WNK4 regulates Na+ channel and K+ channel activity and has implications for aldosterone signaling and K+ homeostasis". Proceedings of the National Academy of Sciences of the United States of America. 104 (10): 4025–9. doi:10.1073/pnas.0611728104. PMID 17360471.
- Fu Y, Subramanya A, Rozansky D, Cohen DM (June 2006). "WNK kinases influence TRPV4 channel function and localization". American Journal of Physiology. Renal Physiology. 290 (6): F1305-14. doi:10.1152/ajprenal.00391.2005. PMID 16403833.
- Jiang Y, Ferguson WB, Peng JB (February 2007). "WNK4 enhances TRPV5-mediated calcium transport: potential role in hypercalciuria of familial hyperkalemic hypertension caused by gene mutation of WNK4". American Journal of Physiology. Renal Physiology. 292 (2): F545-54. doi:10.1152/ajprenal.00187.2006. PMID 17018846.
- Murthy M, Cope G, O'Shaughnessy KM (October 2008). "The acidic motif of WNK4 is crucial for its interaction with the K channel ROMK". Biochemical and Biophysical Research Communications. 375 (4): 651–4. doi:10.1016/j.bbrc.2008.08.076. PMID 18755144.
- Ponce-Coria J, Markadieu N, Austin TM, Flammang L, Rios K, Welling PA, Delpire E (June 2014). "A novel Ste20-related proline/alanine-rich kinase (SPAK)-independent pathway involving calcium-binding protein 39 (Cab39) and serine threonine kinase with no lysine member 4 (WNK4) in the activation of Na-K-Cl cotransporters". The Journal of Biological Chemistry. 289 (25): 17680–8. doi:10.1074/jbc.M113.540518. PMID 24811174.
- Brooks AM, Owens M, Sayer JA, Salzmann M, Ellard S, Vaidya B (August 2012). "Pseudohypoaldosteronism type 2 presenting with hypertension and hyperkalaemia due to a novel mutation in the WNK4 gene". QJM. 105 (8): 791–4. doi:10.1093/qjmed/hcr119. PMID 21764813.
- Gong H, Tang Z, Yang Y, Sun L, Zhang W, Wang W, et al. (June 2008). "A patient with pseudohypoaldosteronism type II caused by a novel mutation in WNK4 gene". Endocrine. 33 (3): 230–4. doi:10.1007/s12020-008-9084-8. PMID 19016006. S2CID 45040268.
- Golbang AP, Murthy M, Hamad A, Liu CH, Cope G, Van't Hoff W, et al. (August 2005). "A new kindred with pseudohypoaldosteronism type II and a novel mutation (564D>H) in the acidic motif of the WNK4 gene". Hypertension. 46 (2): 295–300. doi:10.1161/01.HYP.0000174326.96918.d6. PMID 15998707.
- Zhang C, Wang Z, Xie J, Yan F, Wang W, Feng X, et al. (2011). "Identification of a novel WNK4 mutation in Chinese patients with pseudohypoaldosteronism type II". Nephron Physiology. 118 (3): 53–61. doi:10.1159/000321879. PMID 21196779. S2CID 11164311.
- Na T, Wu G, Zhang W, Dong WJ, Peng JB (January 2013). "Disease-causing R1185C mutation of WNK4 disrupts a regulatory mechanism involving calmodulin binding and SGK1 phosphorylation sites". American Journal of Physiology. Renal Physiology. 304 (1): F8–F18. doi:10.1152/ajprenal.00284.2012. PMC 3543615. PMID 23054253.
- San-Cristobal P, Pacheco-Alvarez D, Richardson C, Ring AM, Vazquez N, Rafiqi FH, et al. (March 2009). "Angiotensin II signaling increases activity of the renal Na-Cl cotransporter through a WNK4-SPAK-dependent pathway". Proceedings of the National Academy of Sciences of the United States of America. 106 (11): 4384–9. doi:10.1073/pnas.0813238106. PMID 19240212.
- Castañeda-Bueno M, Cervantes-Pérez LG, Vázquez N, Uribe N, Kantesaria S, Morla L, et al. (May 2012). "Activation of the renal Na+:Cl- cotransporter by angiotensin II is a WNK4-dependent process". Proceedings of the National Academy of Sciences of the United States of America. 109 (20): 7929–34. doi:10.1073/pnas.1200947109. PMID 22550170.
- Chiga M, Rai T, Yang SS, Ohta A, Takizawa T, Sasaki S, Uchida S (December 2008). "Dietary salt regulates the phosphorylation of OSR1/SPAK kinases and the sodium chloride cotransporter through aldosterone". Kidney International. 74 (11): 1403–9. doi:10.1038/ki.2008.451. PMID 18800028.
- Rozansky DJ, Cornwall T, Subramanya AR, Rogers S, Yang YF, David LL, et al. (September 2009). "Aldosterone mediates activation of the thiazide-sensitive Na-Cl cotransporter through an SGK1 and WNK4 signaling pathway". The Journal of Clinical Investigation. 119 (9): 2601–12. doi:10.1172/JCI38323. PMID 19690383.
- Boyden LM, Choi M, Choate KA, Nelson-Williams CJ, Farhi A, Toka HR, et al. (January 2012). "Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities". Nature. 482 (7383): 98–102. doi:10.1038/nature10814. PMC 3278668. PMID 22266938.
- Louis-Dit-Picard H, Barc J, Trujillano D, Miserey-Lenkei S, Bouatia-Naji N, Pylypenko O, et al. (March 2012). "KLHL3 mutations cause familial hyperkalemic hypertension by impairing ion transport in the distal nephron". Nature Genetics. 44 (4): 456–60, S1-3. doi:10.1038/ng.2218. PMID 22406640. S2CID 112724.
- Wakabayashi M, Mori T, Isobe K, Sohara E, Susa K, Araki Y, et al. (March 2013). "Impaired KLHL3-mediated ubiquitination of WNK4 causes human hypertension". Cell Reports. 3 (3): 858–68. doi:10.1016/j.celrep.2013.02.024. PMID 23453970.