Paclitaxel
Paclitaxel (PTX), sold under the brand name Taxol among others, is a chemotherapy medication used to treat a number of types of cancer.[3] This includes ovarian cancer, esophageal cancer, breast cancer, lung cancer, Kaposi sarcoma, cervical cancer, and pancreatic cancer.[3] It is given by injection into a vein.[3] There is also an albumin-bound formulation.[3]
 | |
 | |
| Clinical data | |
|---|---|
| Trade names | Taxol, others |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a607070 |
| License data |
|
| Pregnancy category |
|
| Routes of administration | Intravenous (IV) |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | 6.5% (by mouth)[2] |
| Protein binding | 89 to 98% |
| Metabolism | Liver (CYP2C8 and CYP3A4) |
| Elimination half-life | 5.8 hours |
| Excretion | Fecal and urinary |
| Identifiers | |
| |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| PDB ligand | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.127.725 |
| Chemical and physical data | |
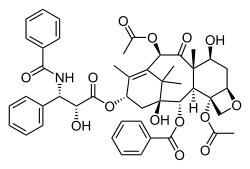
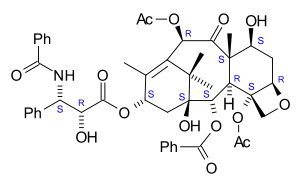
| Formula | C47H51NO14 |
| Molar mass | 853.918 g·mol−1 |
| 3D model (JSmol) | |
| |
| |
| | |
Common side effects include hair loss, bone marrow suppression, numbness, allergic reactions, muscle pains, and diarrhea.[3] Other serious side effects include heart problems, increased risk of infection, and lung inflammation.[3] There are concerns that use during pregnancy may cause birth defects.[4][3] Paclitaxel is in the taxane family of medications.[5] It works by interference with the normal function of microtubules during cell division.[3]
Paclitaxel was first isolated in 1971 from the Pacific yew and approved for medical use in 1993.[6][7] It is on the World Health Organization's List of Essential Medicines.[8] It has been made from precursors, and more recently through cell culture.[7]
Medical use
Paclitaxel is approved in the UK for ovarian, breast, lung, bladder, prostate, melanoma, esophageal, and other types of solid tumor cancers as well as Kaposi's sarcoma.[9]
It is recommended in National Institute for Health and Care Excellence (NICE) guidance of June 2001 that it should be used for non-small-cell lung cancer in patients unsuitable for curative treatment, and in first-line and second-line treatment of ovarian cancer. In September 2001, NICE recommended paclitaxel should be available for the treatment of advanced breast cancer after the failure of anthracyclic chemotherapy, but that its first-line use should be limited to clinical trials. In September 2006, NICE recommended paclitaxel should not be used in the adjuvant treatment of early node-positive breast cancer.[10] In 2018, it is approved in the United States for the treatment of breast, pancreatic, ovarian, Kaposi's sarcoma and non-small-cell lung cancers.[11][12]
Similar compounds
Albumin-bound paclitaxel (trade name Abraxane, also called nab-paclitaxel) is an alternative formulation where paclitaxel is bound to albumin nanoparticles. Much of the clinical toxicity of paclitaxel is associated with the solvent Cremophor EL in which it is dissolved for delivery.[13]
Abraxis BioScience developed Abraxane, in which paclitaxel is bonded to albumin as an alternative delivery agent to the often toxic solvent delivery method. This was approved by the FDA in January 2005 for the treatment of breast cancer after failure of combination chemotherapy for metastatic disease or relapse within six months of adjuvant chemotherapy.[14] It has since been approved for locally advanced or metastatic non-small cell lung cancer and metastatic adenocarcinoma of the pancreas as well.[15]
Synthetic approaches to paclitaxel production led to the development of docetaxel. Docetaxel has a similar set of clinical uses to paclitaxel, and it is marketed under the brand name Taxotere.
Taxanes, including paclitaxel, 10-deacetylbaccatin III, baccatin III, paclitaxel C, and 7-epipaclitaxel, have been found in the leaves and shells of hazel.[16] The finding of these compounds in shells, which are considered discarded material and are mass-produced by many food industries, is of interest for the future availability of paclitaxel.
Restenosis
Paclitaxel is used as an antiproliferative agent for the prevention of restenosis (recurrent narrowing) of coronary and peripheral stents; locally delivered to the wall of the artery, a paclitaxel coating limits the growth of neointima (scar tissue) within stents.[17] Paclitaxel drug-eluting stents for coronary artery placement are sold under the trade name Taxus by Boston Scientific in the United States. Paclitaxel drug-eluting stents for femoropopliteal artery placement are also available.
Side effects
Common side effects include nausea and vomiting, loss of appetite, change in taste, thinned or brittle hair, pain in the joints of the arms or legs lasting two to three days, changes in the color of the nails, and tingling in the hands or toes. More serious side effects such as unusual bruising or bleeding, pain, redness or swelling at the injection site, hand-foot syndrome, change in normal bowel habits for more than two days, fever, chills, cough, sore throat, difficulty swallowing, dizziness, shortness of breath, severe exhaustion, skin rash, facial flushing, female infertility by ovarian damage, and chest pain can also occur. Neuropathy may also occur.[3]
Dexamethasone is given prior to paclitaxel infusion to mitigate some of the side effects.
A number of these side effects are associated with the excipient used, Cremophor EL, a polyoxyethylated castor oil, and allergies to cyclosporine, teniposide, and other drugs containing polyoxyethylated castor oil may increase the risk of adverse reactions to paclitaxel.[18]
Mechanism of action

Paclitaxel is one of several cytoskeletal drugs that target tubulin. Paclitaxel-treated cells have defects in mitotic spindle assembly, chromosome segregation, and cell division. Unlike other tubulin-targeting drugs, such as colchicine, that inhibit microtubule assembly, paclitaxel stabilizes the microtubule polymer and protects it from disassembly. Chromosomes are thus unable to achieve a metaphase spindle configuration. This blocks the progression of mitosis and prolonged activation of the mitotic checkpoint triggers apoptosis or reversion to the G0-phase of the cell cycle without cell division.[19][20]
The ability of paclitaxel to inhibit spindle function is generally attributed to its suppression of microtubule dynamics,[21] but other studies have demonstrated that suppression of dynamics occurs at concentrations lower than those needed to block mitosis. At the higher therapeutic concentrations, paclitaxel appears to suppress microtubule detachment from centrosomes, a process normally activated during mitosis.[22] Paclitaxel binds to the beta-tubulin subunits of microtubules.[23]
Chemistry
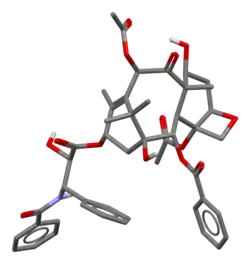
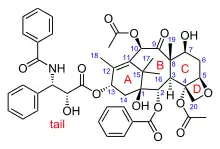
The nomenclature for paclitaxel is structured on a tetracyclic 17-atom skeleton. There are a total of 11 stereocenters. The active stereoisomer is (−)-paclitaxel (shown here).

Production
Bark processing

From 1967 to 1993, almost all paclitaxel produced was derived from bark of the Pacific yew, Taxus brevifolia, the harvesting of which kills the tree in the process.[24] The processes used were descendants of the original isolation method of Monroe Wall and Mansukh Wani; by 1987, the U.S. National Cancer Institute (NCI) had contracted Hauser Chemical Research of Boulder, Colorado, to handle bark on the scale needed for phase II and III trials. While both the size of the wild population of the Pacific yew and the magnitude of the eventual demand for paclitaxel were uncertain, it was clear that an alternative, sustainable source of the natural product would be needed. Initial attempts to broaden its sourcing used needles from the tree, or material from other related Taxus species, including cultivated ones, but these attempts were challenged by the relatively low and often highly variable yields obtained. Early in the 1990s, coincident with increased sensitivity to the ecology of the forests of the Pacific Northwest, paclitaxel was successfully extracted on a clinically useful scale from these sources.[25]
Semisynthesis
Concurrently, synthetic chemists in the U.S. and France had been interested in paclitaxel, beginning in the late 1970s. As noted, by 1992 extensive efforts were underway to accomplish the total synthesis of paclitaxel, efforts motivated by the desire to generate new chemical understanding rather than to achieve practical commercial production. In contrast, the French group of Pierre Potier at the Centre national de la recherche scientifique (CNRS) addressed the matter of overall process yield, showing that it was feasible to isolate relatively large quantities of the compound 10-deacetylbaccatin from the European yew, Taxus baccata, which grew on the CNRS campus and whose needles were available in large quantity. By virtue of its structure, 10-deacetylbaccatin was seen as a viable starting material for a short semisynthesis to produce paclitaxel. By 1988, Poitier and collaborators had published a semisynthetic route from needles of the European yew to paclitaxel.[26]
The view of the NCI, however, was that even this route was not practical. The group of Robert A. Holton had also pursued a practical semisynthetic production route; by late 1989, Holton's group had developed a semisynthetic route to paclitaxel with twice the yield of the Potier process. Florida State University, where Holton worked, signed a deal with Bristol-Myers Squibb (BMS) to license their semisynthesis and future patents. In 1992, Holton patented an improved process with an 80% yield, and BMS took the process in-house and started to manufacture paclitaxel in Ireland from 10-deacetylbaccatin isolated from the needles of the European yew. In early 1993, BMS announced that it would cease reliance on Pacific yew bark by the end of 1995, effectively terminating ecological controversy over its use. This announcement also made good their commitment to develop an alternative supply route, made to the NCI in their cooperative research and development agreement (CRADA) application of 1989.
As of 2013, BMS was using the semisynthetic method utilizing needles from the European yew to produce paclitaxel.[27] Another company which worked with BMS until 2012,[28] Phyton Biotech, Inc., uses plant cell fermentation (PCF) technology.[29] By cultivating a specific Taxus cell line in fermentation tanks, they no longer need ongoing sourcing of material from actual yew tree plantations.[30] Paclitaxel is then captured directly from the suspension broth by a resin allowing concentration to highly enriched powder containing about 40% paclitaxel. The compound is then purified by one chromatographic step followed by crystallization.[31] Compared to the semisynthesis method, PCF eliminates the need for many hazardous chemicals and saves a considerable amount of energy.[32]
In 1993, paclitaxel was discovered as a natural product in a newly described endophytic fungus living in the yew tree.[33] It has since been reported in a number of other endophytic fungi, including Nodulisporium sylviforme, Alternaria taxi, Cladosporium cladosporioides MD2, Metarhizium anisopliae, Aspergillus candidus MD3, Mucor rouxianus, Chaetomella raphigera, Phyllosticta tabernaemontanae, Phomopsis, Pestalotiopsis pauciseta, Phyllosticta citricarpa, Podocarpus sp., Fusarium solani, Pestalotiopsis terminaliae, Pestalotiopsis breviseta, Botryodiplodia theobromae, Gliocladium sp., Alternaria alternata var. monosporus, Cladosporium cladosporioides, Nigrospora sp., Pestalotiopsis versicolor, and Taxomyces andreanae. However, there has been contradictory evidence for its production by endophytes, with other studies finding independent production is unlikely.[34][35]
Biosynthesis
The core synthetic route is via a terpenoid pathway, parts of which having been successfully transplanted into producing strains of E. coli[36] and yeasts.[37]
Total synthesis
By 1992, at least thirty academic research teams globally were working to achieve a total synthesis of this natural product, with the synthesis proceeding from simple natural products and other readily available starting materials.[38] This total synthesis effort was motivated primarily by the desire to generate new chemical understanding, rather than with an expectation of the practical commercial production of paclitaxel. The first laboratories to complete the total synthesis from much less complex starting materials were the research groups of Robert A. Holton, who had the first article to be accepted for publication, and of K. C. Nicolaou who had the first article to appear in print (by a week, on 7 February 1994). Though the Holton submission preceded the Nicolaou by a month (21 December 1993 versus 24 January 1994),[39] the near coincidence of the publications arising from each of these massive, multiyear efforts—11–18 authors appearing on each of the February 1994 publications—has led the ending of the race to be termed a "tie"[40] or a "photo finish",[38] though each group has argued that their synthetic strategy and tactics were superior.[40]
As of 2006, five additional research groups had reported successful total syntheses of paclitaxel: Wender et al. in 1997, and Kuwajima et al. and Mukaiyama et al. in 1998 with further linear syntheses, and Danishefsky et al. in 1996 and Takahashi et al. in 2006 with further convergent syntheses. As of that date, all strategies had aimed to prepare a 10-deacetylbaccatin-type core containing the ABCD ring system, followed generally by last stage addition of the "tail" to the 13-hydroxyl group.[38]
While the "political climate surrounding [paclitaxel] and [the Pacific yew] in the early 1990s [...] helped bolster [a] link between total synthesis and the [paclitaxel] supply problem," and though total synthesis activities were a requisite to explore the structure-activity relationships of paclitaxel via generation of analogs for testing, the total synthesis efforts were never seen "as a serious commercial route" to provide significant quantities of the natural product for medical testing or therapeutic use.[41]
History
The discovery of paclitaxel began in 1962 as a result of a NCI-funded screening program.[7] A number of years later it was isolated from the bark of the Pacific yew, Taxus brevifolia, hence its name "taxol".[7]
The discovery was made by Monroe E. Wall and Mansukh C. Wani at the Research Triangle Institute, Research Triangle Park, North Carolina, in 1971.[42] These scientists isolated the natural product from the bark of the Pacific yew tree, determined its structure and named it "taxol", and arranged for its first biological testing. The compound was then developed commercially by BMS, who had the generic name assigned as "paclitaxel".
Plant screening program
In 1955, the NCI in the United States set up the Cancer Chemotherapy National Service Center (CCNSC) to act as a public screening center for anticancer activity in compounds submitted by external institutions and companies.[43] Although the majority of compounds screened were of synthetic origin, one chemist, Jonathan Hartwell, who was employed there from 1958 onwards, had experience with natural product derived compounds, and began a plant screening operation.[44] After some years of informal arrangements, in July 1960, the NCI commissioned the United States Department of Agriculture (USDA) botanists to collect samples from about 1,000 plant species per year.[45] On 21 August 1962, one of those botanists, Arthur S. Barclay, collected bark from a single Pacific yew tree in a forest north of the town of Packwood, Washington as part of a four-month trip to collect material from over 200 different species. The material was then processed by a number of specialist CCNSC subcontractors, and one of the tree's samples was found to be cytotoxic in a cellular assay on 22 May 1964.[46]
Accordingly, in late 1964 or early 1965, the fractionation and isolation laboratory run by Monroe E. Wall in Research Triangle Park, North Carolina, began work on fresh Taxus samples, isolating the active ingredient in September 1966 and announcing their findings at an April 1967 American Chemical Society meeting in Miami Beach.[47] They named the pure compound taxol in June 1967.[46] Wall and his colleague Wani published their results, including the chemical structure, in 1971.[48]
The NCI continued to commission work to collect more Taxus bark and to isolate increasing quantities of taxol. By 1969, 28 kg (62 lb) of crude extract had been isolated from almost 1,200 kg (2,600 lb) of bark, although this ultimately yielded only 10 g (0.35 oz) of pure material,[49] but for several years, no use was made of the compound by the NCI. In 1975, it was shown to be active in another in vitro system; two years later, a new department head reviewed the data and finally recommended taxol be moved on to the next stage in the discovery process.[50] This required increasing quantities of purified taxol, up to 600 g (21 oz), and in 1977 a further request for 7,000 lb (3,200 kg) of bark was made.
In 1978, two NCI researchers published a report showing taxol was mildly effective in leukaemic mice.[51] In November 1978, taxol was shown to be effective in xenograft studies.[52] Meanwhile, taxol began to be well known in the cell biology, as well as the cancer community, with a publication in early 1979 by Susan B. Horwitz, a molecular pharmacologist at Albert Einstein College of Medicine, showing taxol had a previously unknown mechanism of action involving the stabilization of microtubules. Together with formulation problems, this increased interest from researchers meant that, by 1980, the NCI envisaged needing to collect 20,000 lb (9,100 kg) of bark.[53] Animal toxicology studies were complete by June 1982, and in November NCI applied for the IND necessary to begin clinical trials in humans.[53]
Early clinical trials, supply and the transfer to BMS
Phase I clinical trials began in April 1984, and the decision to start Phase II trials was made a year later.[54] These larger trials needed more bark and collection of a further 12,000 pounds was commissioned, which enabled some phase II trials to begin by the end of 1986. But by then it was recognized that the demand for taxol might be substantial and that more than 60,000 pounds of bark might be needed as a minimum. This unprecedentedly large amount brought ecological concerns about the impact on yew populations into focus for the first time, as local politicians and foresters expressed unease at the program.[55]
The first public report from a phase II trial in May 1988 showed promising effects in melanoma and refractory ovarian cancer.[56] At this point, Gordon Cragg of the NCI's Natural Product Branch calculated the synthesis of enough taxol to treat all the ovarian cancer and melanoma cases in the US would require the destruction of 360,000 trees annually. For the first time, serious consideration was given to the problem of supply.[55] Because of the practical and, in particular, the financial scale of the program needed, the NCI decided to seek association with a pharmaceutical company, and in August 1989, it published a Cooperative Research and Development Agreement (CRADA) offering its current stock and supply from current bark stocks, and proprietary access to the data so far collected, to a company willing to commit to providing the funds to collect further raw material, isolate taxol, and fund a large proportion of clinical trials. In the words of Goodman and Welsh, authors of a substantial scholarly book on taxol, "The NCI was thinking, not of collaboration, ... but of a hand-over of taxol (and its problems)".[55]
Although the offer was widely advertised, only four companies responded to the CRADA, including the American firm Bristol-Myers Squibb (BMS), which was selected as the partner in December 1989. The choice of BMS later became controversial and was the subject of Congressional hearings in 1991 and 1992. While it seems clear the NCI had little choice but to seek a commercial partner, there was also controversy about the terms of the deal, eventually leading to a report by the General Accounting Office in 2003, which concluded the NIH had failed to ensure value for money.[57] In related CRADAs with the USDA and Department of the Interior, Bristol-Myers Squibb was given exclusive first refusal on all Federal supplies of Taxus brevifolia. This exclusive contract lead to some criticism for giving BMS a "cancer monopoly".[58] Eighteen months after the CRADA, BMS filed a new drug application (NDA), which was given FDA approval at the very end of 1992. [55] Although there was no patent on the compound, the provisions of the Waxman-Hatch Act gave Bristol-Myers Squibb five years exclusive marketing rights.
In 1990, BMS applied to trademark the name taxol as Taxol(R). This was controversially approved in 1992. At the same time, paclitaxel replaced taxol as the generic (INN) name of the compound. Critics, including the journal Nature, argued the name taxol had been used for more than two decades and in more than 600 scientific articles and suggested the trademark should not have been awarded and the BMS should renounce its rights to it.[59] BMS argued changing the name would cause confusion among oncologists and possibly endanger the health of patients. BMS has continued to defend its rights to the name in the courts.[60] BMS has also been criticized for misrepresentation by Goodman and Walsh, who quote from a company report saying "It was not until 1971 that ... testing ... enabled the isolation of paclitaxel, initially described as 'compound 17".[61] This quote is, strictly speaking, accurate: the objection seems to be that this misleadingly neglects to explain that it was the scientist doing the isolation who named the compound taxol and it was not referred to in any other way for more than twenty years. Annual sales peaked in 2000, reaching US$1.6 billion; paclitaxel is now available in generic form.
Society and culture
As of 2006, the cost to the NHS per patient in early breast cancer, assuming four cycles of treatment, was about £4000 (approx. $6000).[62]
Research
Caffeine has been speculated to inhibit paclitaxel-induced apoptosis in colorectal cancer cells.[63]
Aside from its direct clinical use, paclitaxel is used extensively in biological and biomedical research as a microtubule stabilizer. In general, in vitro assays involving microtubules, such as motility assays, rely on paclitaxel to maintain microtubule integrity in the absence of the various nucleating factors and other stabilizing elements found in the cell. For example, it is used for in vitro tests of drugs that aim to alter the behavior of microtubule motor proteins, or for studies of mutant motor proteins. Moreover, Paclitaxel has been used in vitro to inhibit insulin fibrillation; in a molar ratio of 10:1 (insulin:paclitaxel), it hindered insulin fibrillation near 70%. Iso-thermal titration calorimetry (ITC) findings indicated a spontaneous tendency of paclitaxel to interact with insulin through hydrogen bonds and van der Waal's forces.[64] Also, the inhibitory role of paclitaxel is attributed to its impact on the colloidal stability of protein solution, as it was observed that paclitaxel inhibited lysozyme fibrillation by inducing the formation of "off-pathway" oligomeric intermediates and increasing the colloidal stability subsequently.[65] Paclitaxel is sometimes used for in vivo studies as well; it can be fed to test organisms, such as fruit flies, or injected into individual cells, to inhibit microtubule disassembly or to increase the number of microtubules in the cell. Paclitaxel induces remyelination in a demyelinating mouse in vivo[66] and inhibits hPAD2 in vitro though its methyl ester side chain.[67] Angiotech Pharmaceuticals Inc. began phase II clinical trials in 1999[68] as a multiple sclerosis treatment but in 2002, reported that the results showed no statistical significance.[69]
In 2016 in vitro multi-drug resistant mouse tumor cells were treated with paclitaxel encased in exosomes. Doses 98% less than common dosing had the same effect. Also, dye-marked exosomes were able to mark tumor cells, potentially aiding in diagnosis.[70][71]
Additional images
 Space-filling model of paclitaxel
Space-filling model of paclitaxel Rotating paclitaxel molecule model
Rotating paclitaxel molecule model Crystal structure of paclitaxel
Crystal structure of paclitaxel Total charge surface of taxol. Minimum energy conformation.
Total charge surface of taxol. Minimum energy conformation.
References
- "Paclitaxel Use During Pregnancy". Drugs.com. 24 January 2019. Retrieved 19 May 2020.
- Peltier S, Oger JM, Lagarce F, Couet W, Benoît JP (June 2006). "Enhanced oral paclitaxel bioavailability after administration of paclitaxel-loaded lipid nanocapsules". Pharmaceutical Research. 23 (6): 1243–50. doi:10.1007/s11095-006-0022-2. PMID 16715372. S2CID 231917.
- "Paclitaxel". The American Society of Health-System Pharmacists. Archived from the original on September 14, 2017. Retrieved January 2, 2015.
- Berveiller P, Mir O (2012). "Taxanes during pregnancy: probably safe, but still to be optimized". Oncology. 83 (4): 239–40. doi:10.1159/000341820. PMID 22907122.
- Chang AE, Ganz PA, Hayes DF, Kinsella T, Pass HI, Schiller JH, Stone RM, Strecher V (2007). Oncology: An Evidence-Based Approach. Springer Science & Business Media. p. 34. ISBN 9780387310565. Archived from the original on 2016-12-21.
- Fischer J, Ganellin CR (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 512. ISBN 9783527607495. Archived from the original on 2016-12-21.
- "Taxol® (NSC 125973)". National Cancer Institute. Archived from the original on 5 September 2015. Retrieved 14 February 2016. Wayback machine
- World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- Saville MW, Lietzau J, Pluda JM, Feuerstein I, Odom J, Wilson WH, et al. (July 1995). "Treatment of HIV-associated Kaposi's sarcoma with paclitaxel". Lancet (Submitted manuscript). 346 (8966): 26–8. doi:10.1016/S0140-6736(95)92654-2. PMID 7603142. S2CID 44624033. Archived from the original on 2019-06-26. Retrieved 2018-10-28.
- "British National Formulary".
- "Paclitaxel, Protein-Bound Suspension". Paclitaxel, Protein-Bound Suspension. Cancer.Org. January 6, 2015. Retrieved January 24, 2015.
- Product Information: TAXOL(R) IV injection, paclitaxel IV injection. Bristol-Myers Squibb Company, Princeton, NJ, 2013. Accessed from https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/020262s051lbl.pdf on 4 October 2018.
- Gelderblom H, Verweij J, Nooter K, Sparreboom A (September 2001). "Cremophor EL: the drawbacks and advantages of vehicle selection for drug formulation". European Journal of Cancer. 37 (13): 1590–8. doi:10.1016/S0959-8049(01)00171-X. PMID 11527683.
- "Abraxane Drug Information Archived 2005-05-26 at the Wayback Machine." Food and Drug Administration. January 7, 2005. Retrieved on March 9, 2007.
- Product Information: ABRAXANE(R) intravenous injection suspension, paclitaxel protein-bound particles intravenous injection suspension. Celgene Corporation (per FDA), Summit, NJ, 2018. Accessed from https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/021660s045lbl.pdf on 4 October 2018.
- Ottaggio L, Bestoso F, Armirotti A, Balbi A, Damonte G, Mazzei M, et al. (January 2008). "Taxanes from Shells and Leaves of Corylus avellana". Journal of Natural Products. 71 (1): 58–60. doi:10.1021/np0704046. PMID 18163585.
- Heldman AW, Cheng L, Jenkins GM, Heller PF, Kim DW, Ware M, et al. (May 2001). "Paclitaxel stent coating inhibits neointimal hyperplasia at 4 weeks in a porcine model of coronary restenosis". Circulation. 103 (18): 2289–95. doi:10.1161/01.CIR.103.18.2289. PMID 11342479.
- "Medline Plus Entry for Paclitaxel Injection Archived 2010-02-12 at the Wayback Machine." MEDLINE. Last Reviewed 09/01/2008. Accessed 10-2-21.
- Bharadwaj R, Yu H (March 2004). "The spindle checkpoint, aneuploidy, and cancer". Oncogene. 23 (11): 2016–27. doi:10.1038/sj.onc.1207374. PMID 15021889.
- Brito DA, Yang Z, Rieder CL (August 2008). "Microtubules do not promote mitotic slippage when the spindle assembly checkpoint cannot be satisfied". The Journal of Cell Biology. 182 (4): 623–9. doi:10.1083/jcb.200805072. PMC 2518701. PMID 18710927.
- Jordan MA, Wilson L (April 2004). "Microtubules as a target for anticancer drugs". Nature Reviews. Cancer. 4 (4): 253–65. doi:10.1038/nrc1317. PMID 15057285. S2CID 10228718.
- Ganguly A, Yang H, Cabral F (November 2010). "Paclitaxel-dependent cell lines reveal a novel drug activity". Molecular Cancer Therapeutics. 9 (11): 2914–23. doi:10.1158/1535-7163.MCT-10-0552. PMC 2978777. PMID 20978163.
- Löwe J, Li H, Downing KH, Nogales E (November 2001). "Refined structure of alpha beta-tubulin at 3.5 A resolution". Journal of Molecular Biology. 313 (5): 1045–57. doi:10.1006/jmbi.2001.5077. PMID 11700061.
- Gersmann H, Aldred J (10 November 2011). "Medicinal tree used in chemotherapy drug faces extinction". The Guardian. Archived from the original on 16 February 2017. Retrieved 2017-02-15.
- Goodman & Walsh 2001, pp. 172–5.
- Goodman & Walsh 2001, pp. 100–1.
- "Paclitaxel Injection, USP" (PDF). Injectable Pharmaceuticals. Archived from the original (PDF) on 2016-09-18. Retrieved 2016-04-22.
- "History". Archived from the original on 24 May 2016. Retrieved 22 April 2016.
- "Phyton BioTech Paclitaxel". Archived from the original on 7 August 2016. Retrieved 22 April 2016.
- Imseng N, Schillberg S, Schürch C, Schmid D, Schütte K, Gorr G, Eibl D, Eibl R (2014). Meyer H, Schmidhalter D (eds.). Suspension Culture of Plant Cells under Heterotrophic Conditions. Industrial Scale Suspension Culture of Living Cells. Wiley-Blackwell. pp. 224–257. ISBN 978-3-527-33547-3.
- Gilbert Gorr and Roland Franke. Commercial Pharmaceutical Production of Complex APIs via Plant Cell Fermentation (PCF®) Technology. Presentation at CPhI 2015, Oct. 13th.
- "2004 Greener Synthetic Pathways Award: Bristol-Myers Squibb Company: Development of a Green Synthesis for TAXOL Manufacture via Plant Cell Fermentation and Extraction". Archived from the original on 2006-10-02.
- Stierle A, Strobel G, Stierle D (April 1993). "Taxol and taxane production by Taxomyces andreanae, an endophytic fungus of Pacific yew". Science. 260 (5105): 214–6. Bibcode:1993Sci...260..214S. doi:10.1126/science.8097061. PMID 8097061.
- Staniek A, Woerdenbag HJ, Kayser O (December 2009). "Taxomyces andreanae: a presumed paclitaxel producer demystified?". Planta Medica. 75 (15): 1561–6. doi:10.1055/s-0029-1186181. PMID 19809969.
- Heinig U, Scholz S, Jennewein S (2013). "Getting to the bottom of taxol biosynthesis by fungi" (PDF). Fungal Diversity. 60: 161–170. doi:10.1007/s13225-013-0228-7. S2CID 18642421.
- Boghigian BA, Myint M, Wu J, Pfeifer BA (November 2011). "Simultaneous production and partitioning of heterologous polyketide and isoprenoid natural products in an Escherichia coli two-phase bioprocess". Journal of Industrial Microbiology & Biotechnology. 38 (11): 1809–20. doi:10.1007/s10295-011-0969-9. PMID 21487833. S2CID 6538645.
- Engels B, Dahm P, Jennewein S (2008). "Metabolic engineering of taxadiene biosynthesis in yeast as a first step towards Taxol (Paclitaxel) production". Metabolic Engineering. 10 (3–4): 201–6. doi:10.1016/j.ymben.2008.03.001. PMID 18485776.
- Hall N (2003). "Creating complexity: The beauty and logic of synthesis". Chem. Commun (6): 661–664. doi:10.1039/b212248k. PMID 12703766.
- See N. Hall, ibid. See also the American Chemical Society publication Chemical and Engineering News (C&EN), Feb. 21, 1994, page 32, and primary citations appearing at Holton and Nicolaou taxol total synthesis articles.
- Flam F (February 1994). "Race to synthesize taxol ends in a tie". Science. 263 (5149): 911. Bibcode:1994Sci...263..911F. doi:10.1126/science.7906053. PMID 7906053.
- Goodman & Walsh 2001, pp. 179–182.
- Wall ME, Wani MC (February 1995). "Camptothecin and taxol: discovery to clinic--thirteenth Bruce F. Cain Memorial Award Lecture". Cancer Research. 55 (4): 753–60. PMID 7850785. Archived from the original on November 24, 2016.
- Goodman & Walsh 2001, p. 17.
- Goodman & Walsh 2001, p. 22.
- Goodman & Walsh 2001, pp. 25,28.
- Goodman & Walsh 2001, p. 51.
- Wall ME, Wani MC (February 1995). "Camptothecin and taxol: discovery to clinic--thirteenth Bruce F. Cain Memorial Award Lecture". Cancer Research. 55 (4): 753–60. PMID 7850785.
- Wani MC, Taylor HL, Wall ME, Coggon P, McPhail AT (May 1971). "Plant antitumor agents. VI. The isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia". Journal of the American Chemical Society. 93 (9): 2325–7. doi:10.1021/ja00738a045. PMID 5553076.
- Goodman & Walsh 2001, p. 81.
- Goodman & Walsh 2001, pp. 79,81.
- Fuchs DA, Johnson RK (August 1978). "Cytologic evidence that taxol, an antineoplastic agent from Taxus brevifolia, acts as a mitotic spindle poison". Cancer Treatment Reports. 62 (8): 1219–22. PMID 688258.
- Goodman & Walsh 2001, p. 95.
- Goodman & Walsh 2001, p. 97
- Goodman & Walsh 2001, p. 115.
- Goodman & Walsh 2001, p. 120
- Rowinsky EK, Donehower RC, Rosenshein NB, Ettinger DS, McGuire WP (1988). "Phase II study of taxol in advanced epithelial malignancies". Proceedings of the Association of Clinical Oncology. 7: 136.
- "Technology Transfer: NIH-Private Sector Partnership in the Development of Taxol" (PDF). Archived from the original (PDF) on 2007-07-26. Retrieved 2016-07-17.
- Nader R, Love J (February 1993). "Looting the medicine chest: how Bristol-Myers Squibb made off with the public's cancer research". The Progressive. Archived from the original on 2004-09-24.
- "Names for hi-jacking". Nature. 373 (6513): 370. February 1995. Bibcode:1995Natur.373..370.. doi:10.1038/373370a0. PMID 7830775. S2CID 31510966.
- Goodman & Walsh 2001, p. 170.
- Bristol-Myers Squibb, The development of TAXOL (paclitaxel), March 1997, as cited in Goodman & Walsh 2001, p. 2
- "NICE Guidance TA108". Archived from the original on 2007-06-30.
- Mhaidat NM, Alzoubi KH, Al-Azzam SI, Alsaad AA (January 2014). "Caffeine inhibits paclitaxel‑induced apoptosis in colorectal cancer cells through the upregulation of Mcl‑1 levels". Molecular Medicine Reports. 9 (1): 243–8. doi:10.3892/mmr.2013.1763. PMID 24173825. Archived from the original on 2015-06-22.
- Kachooei E, Moosavi-Movahedi AA, Khodagholi F, Mozaffarian F, Sadeghi P, Hadi-Alijanvand H, et al. (June 2014). "Inhibition study on insulin fibrillation and cytotoxicity by paclitaxel". Journal of Biochemistry. 155 (6): 361–73. doi:10.1093/jb/mvu012. PMID 24535601.
- Kachooei E, Mozaffarian F, Khodagholi F, Sadeghi P, Karami L, Ghasemi A, et al. (May 2018). "Paclitaxel inhibited lysozyme fibrillation by increasing colloidal stability through formation of "off-pathway" oligomers". International Journal of Biological Macromolecules. 111: 870–879. doi:10.1016/j.ijbiomac.2018.01.072. PMID 29352977.
- Moscarello MA, Mak B, Nguyen TA, Wood DD, Mastronardi F, Ludwin SK (April 2002). "Paclitaxel (Taxol) attenuates clinical disease in a spontaneously demyelinating transgenic mouse and induces remyelination". Multiple Sclerosis. 8 (2): 130–8. doi:10.1191/1352458502ms776oa. PMID 11990870. S2CID 45994154.
- Musse AA, Polverini E, Raijmakers R, Harauz G (October 2008). "Kinetics of human peptidylarginine deiminase 2 (hPAD2)--reduction of Ca2+ dependence by phospholipids and assessment of proposed inhibition by paclitaxel side chains". Biochemistry and Cell Biology = Biochimie et Biologie Cellulaire. 86 (5): 437–47. doi:10.1139/o08-124. PMID 18923545.
- MS Society of Canada Phase II Clinical trial of Micellar Paclitaxel for secondary-progressive MS underway in Canada Archived 2012-03-15 at the Wayback Machine
- MS Society of Canada Angiotech Halts Study of Micellar Paclitaxel stating no benefit of statistical significance seen Archived 2012-03-15 at the Wayback Machine
- Lavars, Nick (2016-01-14). "Cloaking chemo drugs in cellular bubbles destroys cancer with one fiftieth of a regular dose". www.gizmag.com. Archived from the original on 2016-02-24. Retrieved 2016-02-14.
- Kim MS, Haney MJ, Zhao Y, Mahajan V, Deygen I, Klyachko NL, et al. (April 2016). "Development of exosome-encapsulated paclitaxel to overcome MDR in cancer cells". Nanomedicine. 12 (3): 655–664. doi:10.1016/j.nano.2015.10.012. PMC 4809755. PMID 26586551.
Further reading
- Goodman J, Walsh V (5 March 2001). The Story of Taxol: Nature and Politics in the Pursuit of an Anti-Cancer Drug. Cambridge University Press. ISBN 978-0-521-56123-5.
External links
- "Paclitaxel". Drug Information Portal. U.S. National Library of Medicine.
- "Paclitaxel". National Cancer Institute. 5 October 2006.
- "Paclitaxel". NCI Drug Dictionary. 2 February 2011.
- Molecule of the Month: TAXOL by Neil Edwards, University of Bristol.
- A Tale of Taxol from Florida State University.
- Berenson, Alex (October 1, 2006). "Hope, at $4,200 a Dose". The New York Times. Retrieved 2007-03-31.
| Authority control |
|---|